
Análisis transcriptómico comparativo en células infectadas y no infectadas por Senecavirus A

Análisis transcriptómico comparativo en células infectadas y no infectadas por Senecavirus A
 Yan Li1,2†Huanhuan Chu1,3†Yujia Jiang1,4Ziwei Li1Jie Wang1
Yan Li1,2†Huanhuan Chu1,3†Yujia Jiang1,4Ziwei Li1Jie Wang1- 1Facultad de Medicina Veterinaria, Universidad Agrícola de Qingdao, Qingdao, China
- número arábigoCentro Qingdao para el Control y la Prevención de Enfermedades Animales, Qingdao, China
- 3Facultad de Medicina Veterinaria, Universidad Northwest A&F, Yangling, China
- 4Qingdao Zhongren-OLand Bioengineering Co., Ltd., Qingdao, China
El Senecavirus A (SVA) es un virus emergente que causa la enfermedad vesicular en cerdos, clínicamente indistinguible de otras enfermedades vesiculares de alta consecuencia. Este virus pertenece al género Senecavirus de la familia Picornaviridae. Su genoma es un ARN monocatenario de sentido positivo, de aproximadamente 7.300 nt de longitud, con una cola de poli(A) de 3′ pero sin una estructura con tapa de 5′ extremos. El SVA puede propagarse eficientemente en diferentes células, incluidas algunas líneas celulares no derivadas de cerdos. Un SVA de tipo salvaje fue rescatado previamente de su clon de ADNc utilizando genética inversa en nuestro laboratorio. En el presente estudio, la línea celular BSR-T7/5 fue inoculada con el SVA de paso-5. A las 12 h después de la inoculación, se recolectaron células infectadas y no infectadas por SVA de forma independiente para el análisis en transcriptómica comparativa. Los resultados mostraron un total de 628 genes expresados diferencialmente, incluidos 565 regulados al alza y 63 regulados a la baja, lo que sugiere que la infección por SVA estimuló significativamente el inicio de la transcripción en las células. Los análisis de enriquecimiento de GO y KEGG demostraron que el SVA ejercía múltiples efectos sobre las vías relacionadas con la inmunidad en las células. Además, los datos de secuenciación de ARN se sometieron a otros análisis en profundidad, como el polimorfismo de un solo nucleótido, los factores de transcripción y las interacciones proteína-proteína. El presente estudio, junto con nuestras investigaciones previas de proteómica y metabolómica, proporciona una visión multiómica de la interacción entre SVA y sus huéspedes.
1 Introducción
Se ha demostrado que el Senecavirus A (SVA), como virus emergente, es un agente causal de enfermedad vesicular en cerdos (1-5). Los cerdos infectados por SVA desarrollan lesiones vesiculares principalmente en el hocico, el espolón o la banda coronaria. Otros signos incluyen cojera, anorexia, letargo, hiperemia cutánea y fiebre (6, 7). Los signos inducidos por el SVA son clínicamente indistinguibles de los de otras enfermedades vesiculares en cerdos (8). El brote de infección por SVA se ha notificado recientemente en varios países, entre ellos Canadá, Estados Unidos, Brasil, China, Tailandia, Vietnam y Chile. El riesgo de transmisión del SVA ha atraído una gran atención de la industria porcina de todo el mundo.
SVA es el único miembro del género Senecavirus de la familia Picornaviridae (9). El virión es una partícula icosaédrica típica sin envoltura. Alberga un genoma de ARN monocatenario de sentido positivo, de aproximadamente 7.300 nt de longitud, compuesto por una región no traducida (UTR) 5′, una región codificante larga y una UTR 3′. Al igual que los de otros picornavirus, el extremo 5′ del genoma del SVA no contiene una estructura de tapa. Por el contrario, un péptido corto (VPg) está unido covalentemente al extremo 5′ y desempeña un papel esencial en la síntesis del genoma del SVA. La UTR 5′ tiene un sitio de entrada del ribosoma interno de tipo IV (10), estructural y funcionalmente similar a los de los pestivirus (11), lo que permite el inicio de la traducción de poliproteínas de manera independiente de la tapa. La UTR 3′ tiene aproximadamente 70 nt de longitud, seguida de una cola de poli(A) de longitud variable (12). La región codificante de la poliproteína SVA sigue el diseño estándar «L-VP4-VP2-VP3-VP1-2A-2B-2C-3A-3B-3C-3D». Después de la infección por SVA, la poliproteína viral se traducirá en el citoplasma y luego se dividirá gradualmente en 12 proteínas, a saber, L, VP4, VP2, VP3, VP1, 2A, 2B, 2C, 3A, 3B, 3C y 3D (13). Las VP4, VP2, VP3 y VP1 como proteínas estructurales participan en la morfogénesis viral. Las otras son proteínas no estructurales, aunque no involucradas en el paquete de viriones, que desempeñan un papel crucial en la replicación viral (14-16).
La secuenciación de ARN (RNA-seq) es una técnica que utiliza la secuenciación de nueva generación para revelar la presencia y la cantidad de moléculas de ARN en una muestra biológica, proporcionando una instantánea de la expresión génica en la muestra, también conocida como transcriptoma. Un transcriptoma es la gama completa de moléculas de ARNm expresadas por un organismo. La técnica RNA-seq contribuye a identificar un transcriptoma en una población determinada, incluso en una sola célula (17). La transcriptómica comparativa facilita la elucidación de la diferenciación entre dos grupos (poblaciones, especies, etc.) en sus transcripciones alternativas de empalme de genes, modificaciones postranscripcionales, fusión de genes, polimorfismo de un solo nucleótido (SNP) y cambios en la expresión génica a lo largo del tiempo (18). Los virus de ADN grandes, como el citomegalovirus humano y el virus de la peste porcina africana, contenían genomas muy largos. Cada uno de estos virus tiene un transcriptoma complicado (19, 20) en las células infectadas por virus. Por el contrario, algunos virus de ARN pequeños, como el picornavirus, solo tienen un «transcriptoma» simple, es decir, un solo genoma de ARN. Por lo tanto, no tiene sentido descubrir un «transcriptoma» de picornavirus solo basado en un picornavirus dado.
El SVA puede desencadenar una variedad de cambios metabólicos y bioquímicos en las células a través de mecanismos específicos o inespecíficos del virus (21-23). Por ejemplo, la proteína SVA 2C puede dirigirse a las mitocondrias y provocar la liberación de citocromo C en el citoplasma, que activa las caspasas-9 y -3 en una serie de cascadas de señalización para inducir la aparición de la apoptosis (24). Además, la infección por SVA es capaz de afectar el nivel de transcripción en los huéspedes. Por ejemplo, la proteína SVA 2C puede bloquear la transcripción del gen 56 estimulado por interferón y el β de interferón para debilitar la inmunidad innata en los huéspedes (21). Hemos demostrado que la infección por SVA puede conducir a cambios significativos en el proteoma celular y el metaboloma, incluso en una etapa temprana de la infección (25, 26). La diferenciación del proteoma celular causada por el virus está estrechamente relacionada con el cambio en el transcriptoma celular. Por lo tanto, se llevó a cabo un análisis transcriptómico comparativo para descubrir un perfil de cambios inducidos por SVA en el transcriptoma celular en la etapa temprana de la infección.
2 Materiales y métodos
2.1 Línea celular y virus
La línea celular BSR-T7/5, derivada de la célula de riñón del hámster bebé, fue amablemente proporcionada por el Centro de Salud Animal y Epidemiología de China. Esta línea celular se cultivó a 37°C con 5% de CO2 en medio de Eagle modificado de Dulbecco (DMEM), suplementado con 4% de suero fetal bovino (VivaCell, Shanghai, China), penicilina (100 U/mL), estreptomicina (100 μg/mL) y anfotericina B (0,25 μg/mL). El SVA de tipo salvaje fue rescatado previamente de un clon de ADNc de longitud completa (27), derivado genéticamente de un aislado de SVA, CH-LX-01-2016 (28).
2.2 Preparación de la muestra
Las células BSR-T7/5 se sembraron en seis matraces T25 para su cultivo a 37 °C. Cuando las células estaban confluentes en un 90%, se seleccionaron aleatoriamente tres matraces para la incubación con el SVA de paso-5 a un MOI (multiplicidad de infección) de 2,5. Los otros frascos, como controles no infectados, no fueron tratados. Hubo tres muestras infectadas por SVA (S1, S2 y S3) y tres controles no infectados (C1, C2 y C3). Los sobrenadantes se retiraron por separado de los seis frascos a las 12 h después de la inoculación (HPI). Las monocapas celulares se lavaron suavemente con PBS tres veces, seguidas de la extracción de ARN totales utilizando el reactivo TRIzol (Thermo Fisher, Waltham, MA, Estados Unidos), según las instrucciones del fabricante. La concentración, calidad e integridad de los ARN totales se determinaron utilizando el espectrofotómetro NanoDrop (Thermo Fisher, Waltham, MA, Estados Unidos). Se utilizaron tres μg de ARN como material de entrada para preparar la muestra de ARN para cada grupo.
2.3 Análisis de secuenciación de ARN
La preparación de las bibliotecas de secuenciación se llevó a cabo como se describió anteriormente con modificaciones (29). Los ARNm se purificaron a partir de ARN totales utilizando perlas magnéticas unidas a oligonucleótidos poli-T, se fragmentaron aún más y luego se utilizaron como plantillas para producir ADNc. La primera hebra de ADNc se sintetizó utilizando un sistema con cebadores de hexámeros aleatorios y la transcriptasa inversa. La segunda hebra de ADNc se sintetizó a través de la primera hebra con dNTP, solución tampón, ADN polimerasa I y RNasa H. Los salientes restantes se convirtieron en extremos romos a través de actividades de exonucleasa/polimerasa. Después de la adenilación de los extremos 3′ de los fragmentos de ADN, los oligonucleótidos adaptadores de extremos emparejados de Illumina se ligaron para prepararse para la hibridación. Los fragmentos de ADNc de 400 a 500 pb se seleccionaron preferentemente por tamaño utilizando el sistema AMPure XP (Beckman Coulter, Beverly, Estados Unidos). Los fragmentos de ADN con moléculas adaptadoras ligadas en ambos extremos se enriquecieron selectivamente utilizando Illumina PCR Primer Cocktail en una reacción de PCR de 15 ciclos. Los productos se purificaron utilizando el sistema AMPure XP y luego se cuantificaron mediante el ensayo de ADN de alta sensibilidad de Agilent en el Bioanalizador Agilent 2,100 (Agilent Technologies, CA, Estados Unidos). Las librerías de secuenciación fueron sometidas a secuenciación en la plataforma NovaSeq 6.000 (Illumina, CA, Estados Unidos) para la obtención de archivos de imagen.
2.4 Control de calidad y mapeo de lecturas
Los archivos de imagen fueron transformados por el software de la plataforma de secuenciación. Los datos originales se generaron en formato FASTQ (datos sin procesar). Los datos de secuenciación contenían una serie de conectores y lecturas de baja calidad. Se utilizó el software Cutadapt (v1.15) para filtrar los datos de secuenciación (30), obteniendo posteriormente secuencias de alta calidad (datos limpios) para su posterior análisis. Dos genomas de referencia fueron los del hámster dorado (Nº Genbank: PRJNA77669) y el SVA CH-LX-01-2016 (Nº Genbank: KX751945). Las lecturas filtradas se asignaron por separado a ambos genomas de referencia utilizando el programa HISAT2 (v2.0.5) (31).
2.5 Análisis de la expresión diferencial
El análisis de los genes expresados diferencialmente (DEGs) se realizó como se describió anteriormente con modificaciones (32). Se utilizó el HTSeq (v0.9.1) para comparar los valores de recuento de lectura en cada gen como la expresión génica original (33). La expresión génica se estandarizó a través del FPKM (fragmentos por kilobase de transcripción por millón de lecturas mapeadas). Los DEG se determinaron mediante el DESeq (v1.30.0) con los siguientes parámetros de cribado: el cambio de pliegue (FC) > 2 (o <0,5) y el valor significativo de p <0,05 (34). El análisis de agrupamiento bidireccional de todos los DEG se realizó mediante el paquete Pheatmap (v1.0.8). El mapa de calor se dibujó de acuerdo con el nivel de expresión del mismo gen en diferentes grupos y los patrones de expresión de diferentes genes en el mismo grupo, con el método euclidiano para calcular la distancia y el método de ligamiento completo para el agrupamiento.
2.6 Análisis de enriquecimientos de GO y KEGG
Todos los genes fueron mapeados a términos en la base de datos de ontología génica (GO). Se calcularon genes enriquecidos diferencialmente para cada término. El paquete topGO fue diseñado para realizar el análisis de enriquecimiento de GO en los DEG. El valor de p se calculó por el método de distribución hipergeométrica. Se determinó el valor de p <0,05 como estándar de enriquecimiento significativo. Los términos GO se encontraron con genes significativamente enriquecidos diferencialmente, todos los cuales se clasificaron para determinar las principales funciones biológicas. Se utilizó el software ClusterProfiler (v3.4.4) para llevar a cabo el análisis de enriquecimiento de DEGs en las vías KEGG (Kyoto Encyclopedia of Genes and Genomes). Se determinó el valor de p <0,05 como estándar de enriquecimiento significativo (35).
2.7 Otros análisis sobre los datos de RNA-seq
2.7.1 Análisis de nuevas transcripciones
Sobre la base del genoma de referencia existente, se utilizó el software StringTie (http://ccb.jhu.edu/software/stringtie/) para ensamblar las lecturas mapeadas (36). Los resultados del ensamblaje se compararon con las transcripciones conocidas para obtener transcripciones sin anotaciones.
2.7.2 Análisis de eventos de empalme alternativos
Se utilizó el software rMATS (v3.2.5) para descubrir eventos de empalme alternativos (37). Los principales tipos de eventos de empalme alternativos incluyeron el exón omitido (SE), el intrón retenido (RI), el sitio de empalme alternativo 5′ (A5SS), el sitio de empalme alternativo 3′ (A3SS) y los exones mutuamente excluyentes (MXE).
2.7.3 Análisis de sitios SNP
Se utilizó el programa Varscan para obtener los sitios SNP (38). Los criterios de filtrado fueron: (i) la base del sitio SNP Q > 20, (ii) el número de lecturas que cubren el sitio >8, (iii) el número de lecturas que soportan el sitio de mutación >2 y (4) el valor p del locus SNP <0,01.
2.7.4 Predicción de los factores de transcripción
Los factores de transcripción y sus propias familias se predijeron a través de la comparación con la Base de Datos de Factores de Transcripción Animal (AnimalTFDB) (39), una base de datos completa que incluye la clasificación y anotación de factores de transcripción de todo el genoma, cofactores de transcripción y factores de remodelación de la cromatina en numerosos genomas animales.
2.7.5 Análisis de la utilización diferencial de exones
El paquete DEXSeq se utilizó para analizar los datos de RNA-seq para identificar el uso diferencial del exón, como se describió anteriormente (40).
2.7.6 Análisis de interacción en la red de proteínas
Se utilizó la base de datos STRING (https://string-db.org/) para desvelar las supuestas interacciones proteína-proteína (PPI) (41), lo que contribuyó a aclarar la relación entre los genes de interés.
2.8 Validación de la expresión génica mediante RT-qPCR
Se seleccionaron cuatro genes representativos, a saber, el genoma SVA, Nfkbia, Phlda2 y Txnip, para validar el perfil de expresión génica. Se utilizó el gen GAPDH (gliceraldehído-3-fosfato deshidrogenasa) como control interno de referencia. Los cebadores específicos de genes se enumeraron en el archivo suplementario 18 para la validación de RT-qPCR. El análisis de RT-qPCR se realizó con tres repeticiones técnicas, utilizando el AceQ qPCR SYBR Green Master Mix (Vazyme, Nanjing, China) basado en el sistema de PCR en tiempo real LightCycler 480® (Roche, Rotkreuz, Suiza), según las instrucciones del fabricante. Los resultados de la RT-qPCR se analizaron mediante el método 2−ΔΔCt para cuantificar relativamente los cuatro genes de interés (42). Para el análisis estadístico se utilizó el Prisma GraphPad (v8.0) mediante la prueba t de Student de dos colas con corrección de Welch. Los datos se mostraron como medias ± desviaciones estándar de tres experimentos independientes.
3 Resultados
3.1 Secuenciación para el ensamblaje del transcriptoma de novo
Las monocapas celulares BSR-T7/5 no mostraron un efecto citopático (ECP) evidente a 12 hpi (Figura 1). Las células se recolectaron por separado de grupos infectados y no infectados por SVA para extraer ARN totales para la construcción de bibliotecas de ADNc de alta calidad. Los datos primarios relacionados con la biblioteca se enumeran en el Archivo complementario 1. Las bibliotecas de ADNc se sometieron a secuenciación para obtener archivos de imagen, posteriormente transformados en datos brutos para su clasificación estadística, como se muestra en la Tabla 1. Debido a que los datos sin procesar contenían una serie de conectores y lecturas de baja calidad, se utilizó el software Cutadapt (v1.15) para filtrar los datos sin procesar para obtener secuencias limpias de alta calidad, como se indica en el archivo complementario 2. Había tres parámetros clave, a saber, la masa base, el contenido base y la masa media de lecturas. Sus distribuciones se mostraron de forma independiente en los suplementos 3, 4 y 5.

3.2 Mapeo transcriptómico
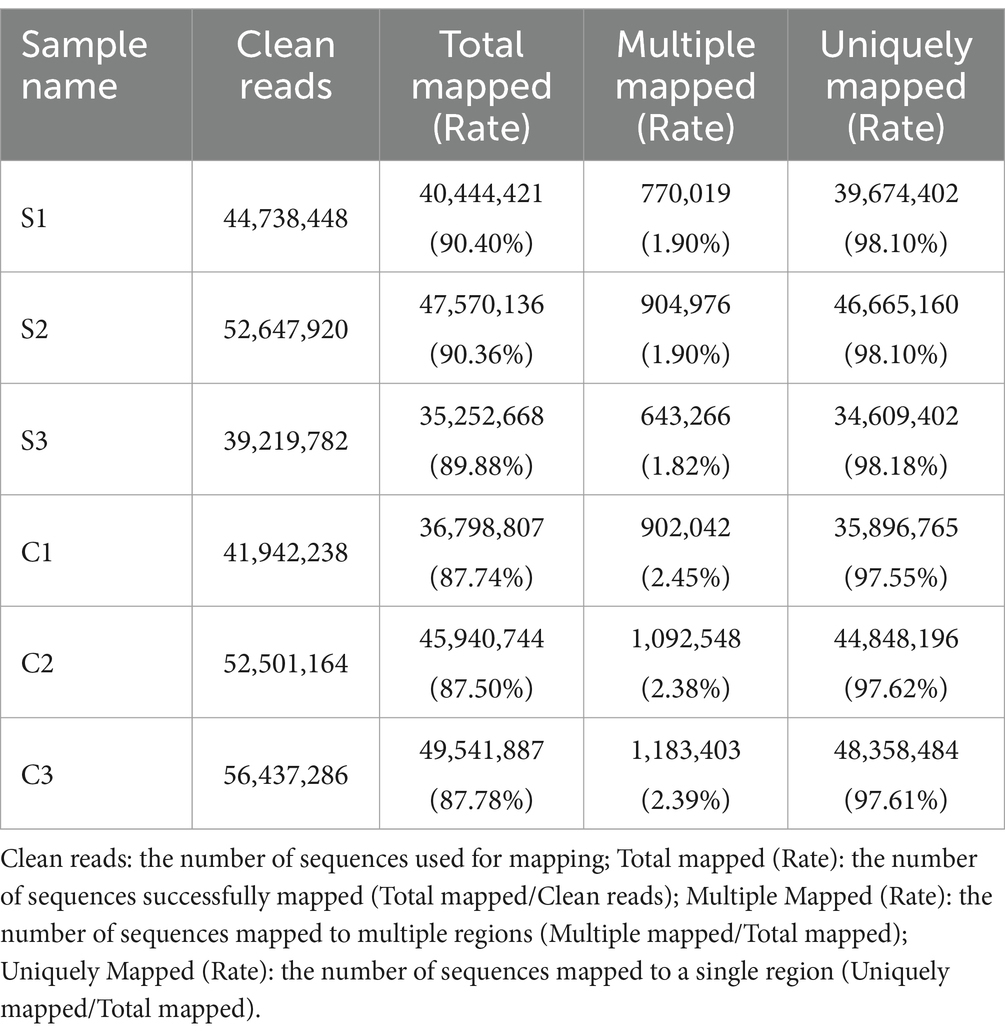
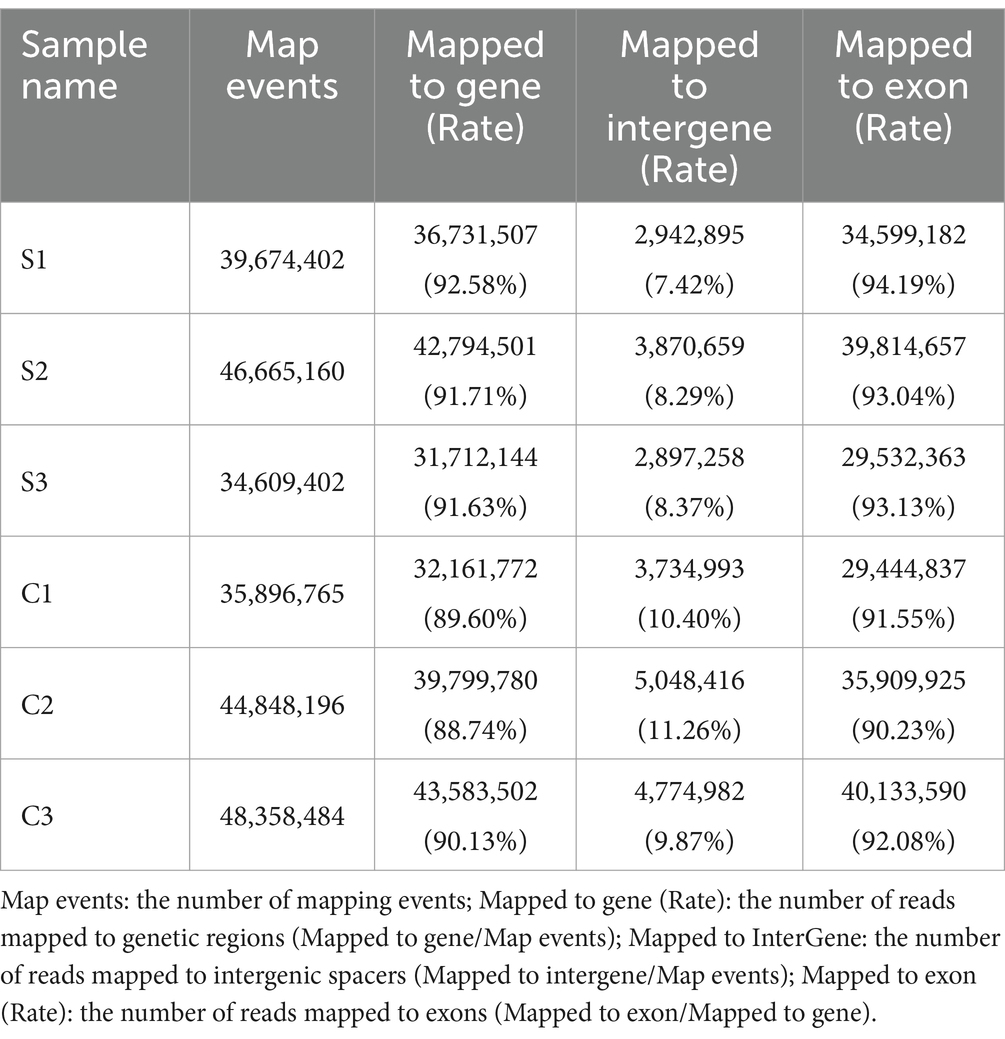
Las lecturas filtradas se mapearon a ambos genomas de referencia, el del hámster dorado y el SVA CH-LX-01-2016, utilizando el programa HISAT2 (v2.0.5). Los resultados del mapeo RNA-seq se enumeraron en la Tabla 2. El perfil global de lecturas se sometió a un análisis estadístico adicional sobre la distribución de lecturas asignadas a ambos genomas, en el que los elementos genéticos incluían la secuencia codificante, el intrón, el espaciador intergénico y la UTR. Los resultados de la cartografía se enumeran en la Tabla 3 y se muestran en el Archivo Suplementario 6. El archivo suplementario 7 exhibió las distribuciones de cobertura de lecturas asignadas a genes. En resumen, aquí se recopiló un conjunto de datos de alta calidad de RNA-Seq, que cumple con un estándar para el análisis bioinformático posterior.
3.3 Perfil de expresión génica
Se identificaron un total de 20.374 genes en los seis grupos (Ficha suplementaria 8), pero estos genes contenían más de 3.000 componentes con un valor de FPKM = 0. FPKM fue un método simple para normalizar los datos de conteo de lecturas, basado en la longitud del gen y el número total de lecturas mapeadas. El nivel de expresión normalizado por FPKM se dividió en diferentes intervalos (Archivo suplementario 9) para los seis grupos, como se muestra en el archivo suplementario 10. El número de genes, ya sea co-identificados en diferentes grupos o reconocidos en un solo grupo, se mostró en la Figura 2A. La distribución de la densidad de los valores de FPKM se mostró en las Figuras 2B, C, como dos formas diferentes. Para validar si la profundidad de secuenciación de RNA-seq era suficiente para el análisis de la expresión génica, se realizó el análisis de saturación para los seis grupos, como se muestra en el archivo suplementario 11. El análisis de correlación de la expresión génica, basado en el cálculo de los coeficientes de correlación de Pearson, se llevó a cabo entre los seis grupos (Figura 2D). Cuanto más cercano era el coeficiente de correlación, mayor era la similitud del patrón de expresión entre los seis grupos. El análisis de componentes principales permitió proyectar un conjunto de datos de alta dimensión en dos o tres dimensiones, como se muestra en la Figura 2E. Cuanto más cercana era la distancia, mayor era la similitud entre los grupos.
3.4 Análisis de la expresión diferencial
Los DEGs se determinaron mediante el DESeq (v1.30.0) con parámetros de cribado, |log2FC| > 1 y el valor significativo de p <0,05. Aquí se identificaron un total de 628 DEG, incluidos 565 componentes regulados al alza (Archivo suplementario 12) y 63 regulados a la baja (Archivo complementario 13). La media base se describió como la «media de los recuentos normalizados de todas las muestras». Los valores medios base de los DEGs, correspondientes a los grupos C y S, se exhibieron en las Figuras 3A, B, respectivamente. Las distribuciones de longitud de secuencia, valor de p y log2FC se exhibieron en las Figuras 3C-H. Un solo asterisco en las Figuras 3G,H indicaba la exclusión del infinito positivo o negativo («Inf» en los archivos suplementarios 12 y 13), respectivamente. La distribución y el grado de expresión diferencial se mostraron gráficamente en un gráfico de volcanes y un mapa de calor, respectivamente.
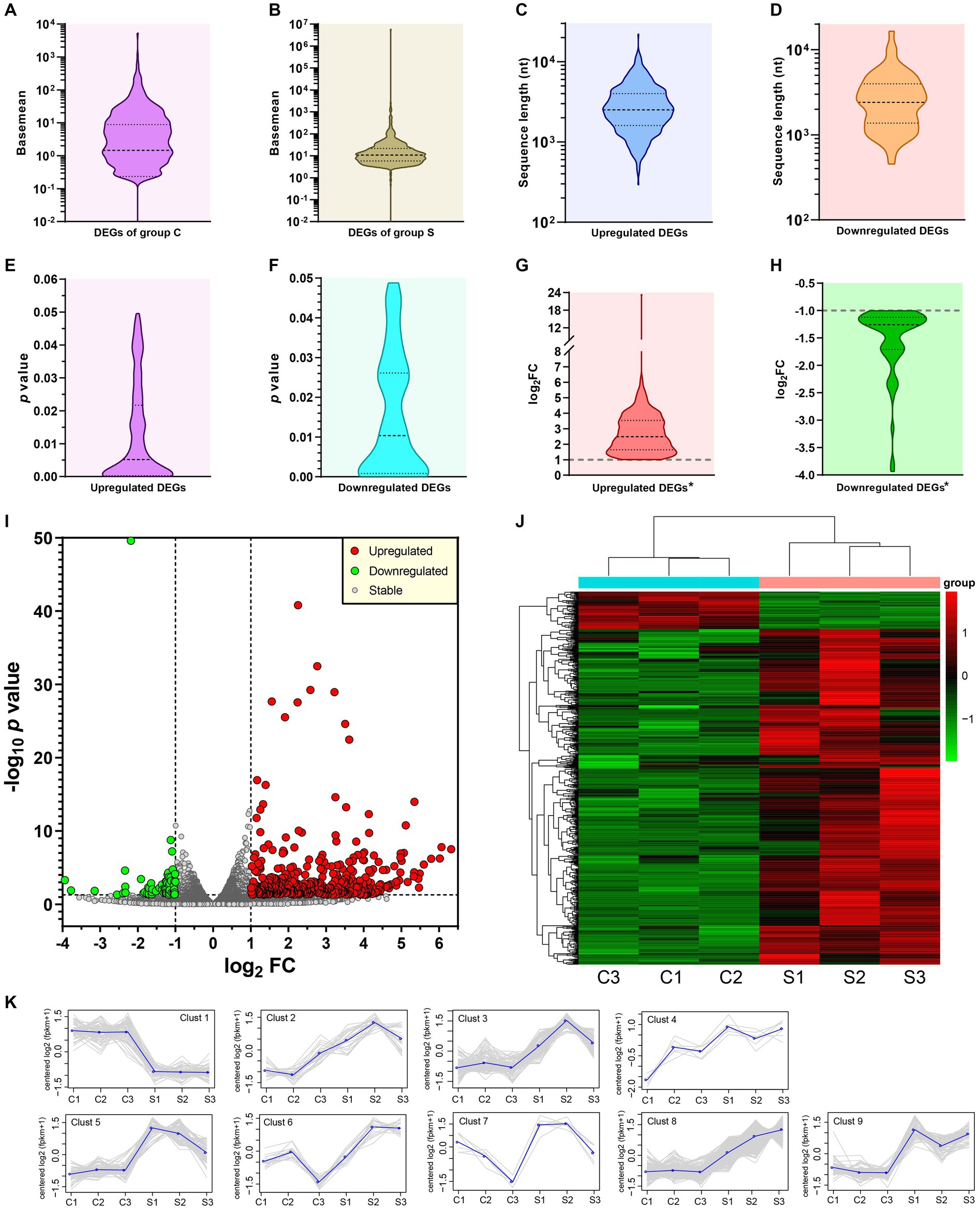
El diagrama del volcán (Figura 3I), dibujado por el software GraphPad Prism, reveló el valor de p frente al FC para todos los genes identificados. Los valores umbral se fijaron como |log2FC| > 1 y valor de p <0,05. Los genes regulados al alza, regulados a la baja y expresados de forma estable se indicaron con círculos rojos, verdes y grises, respectivamente. Se utilizó el paquete de software Pheatmap (1.0.8) del lenguaje R para el análisis de agrupamiento bidireccional para dibujar el mapa de calor (Figura 3J), que proporcionó una representación visual para el agrupamiento jerárquico de los 628 DEG de los seis grupos. Las etiquetas roja y verde representaban los DEG regulados al alza y a la baja, respectivamente. La intensidad del color reflejó el grado de diferenciación en la expresión génica. Los 628 DEG, en función de sus diferencias en los patrones de expresión, se clasificaron en nueve grupos diferentes (Figura 3K), en los que las líneas grises indicaban patrones de expresión, y cada línea azul representaba el valor promedio en cada grupo.
3.5 Análisis de enriquecimiento de GO
El paquete topGO se utilizó para realizar el análisis de enriquecimiento de GO en DEGs. Los términos GO se encontraron con genes significativamente enriquecidos diferencialmente. Los números de las categorías BP, CC y MF fueron 3.093, 357 y 644, respectivamente. Los datos completos de GO se enumeran en el Archivo Suplementario 14 en detalle. En la Figura 4A se muestran los 10 términos de GO estadísticamente significativos para cada categoría de GO. La tasa de falsos descubrimientos (FDR), que osciló entre 0 y 1, se asoció con el grado de enriquecimiento de GO. Cuanto más bajo era el FDR, más significativo era el grado de enriquecimiento. Los términos de GO con los 20 FDR más bajos se mostraron en un diagrama de burbujas (Figura 4B). Cada categoría de GO se organizó como un gráfico acíclico dirigido (Figuras 4C-E), en el que los términos parentales describían categorías funcionales más generales que sus términos de la siguiente generación. Los términos de GO con los 10 FDR más bajos se enmarcaron con rectángulos, y los demás se indicaron con puntos suspensivos. Cuanto más estadísticamente significativo era un término GO, más oscuro era su color.
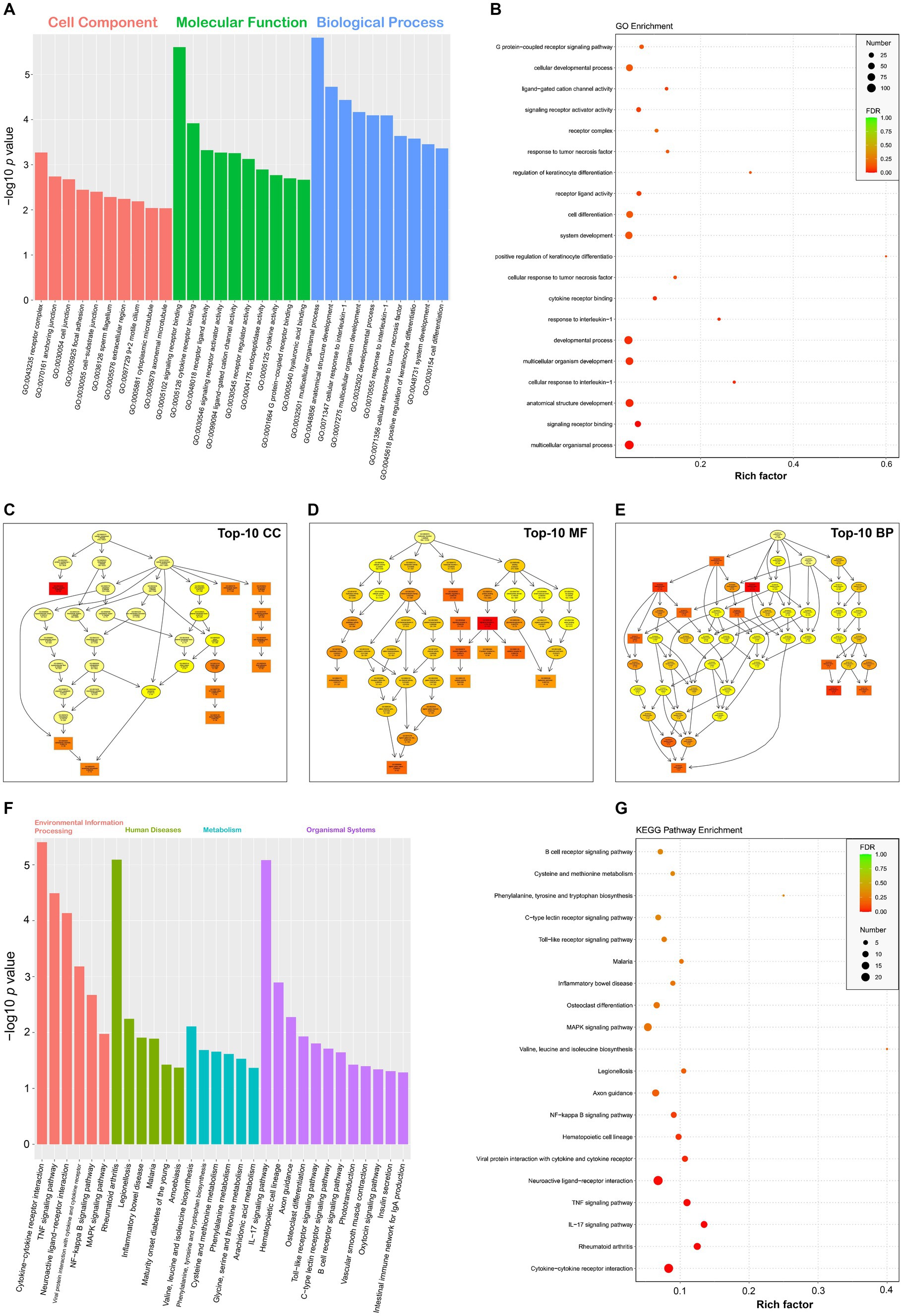
3.6 Análisis de enriquecimiento KEGG
El análisis del enriquecimiento de la vía KEGG se realizó para descubrir las vías relacionadas con DEG. El resultado mostró que los DEG se enriquecieron en un total de 275 vías KEGG (Ficha suplementaria 15). La Figura 4F mostró las 30 principales vías KEGG estadísticamente significativas (valor de p <0,05), clasificadas en cuatro categorías, a saber, procesamiento de información ambiental, enfermedades humanas, metabolismo y sistemas de organismos. De acuerdo con el resultado del enriquecimiento de KEGG, el grado de enriquecimiento se evaluó a través del factor rico, FDR, y el número de DEGs enriquecidos en una vía determinada. Cuanto mayor era el factor rico, más significativo era el grado de enriquecimiento. Cuanto más bajo era el FDR, más significativo era el grado de enriquecimiento. Las vías KEGG con los 20 FDR más bajos se mostraron en un diagrama de burbujas (Figura 4G).
3.7 Otros análisis sobre los datos de RNA-seq
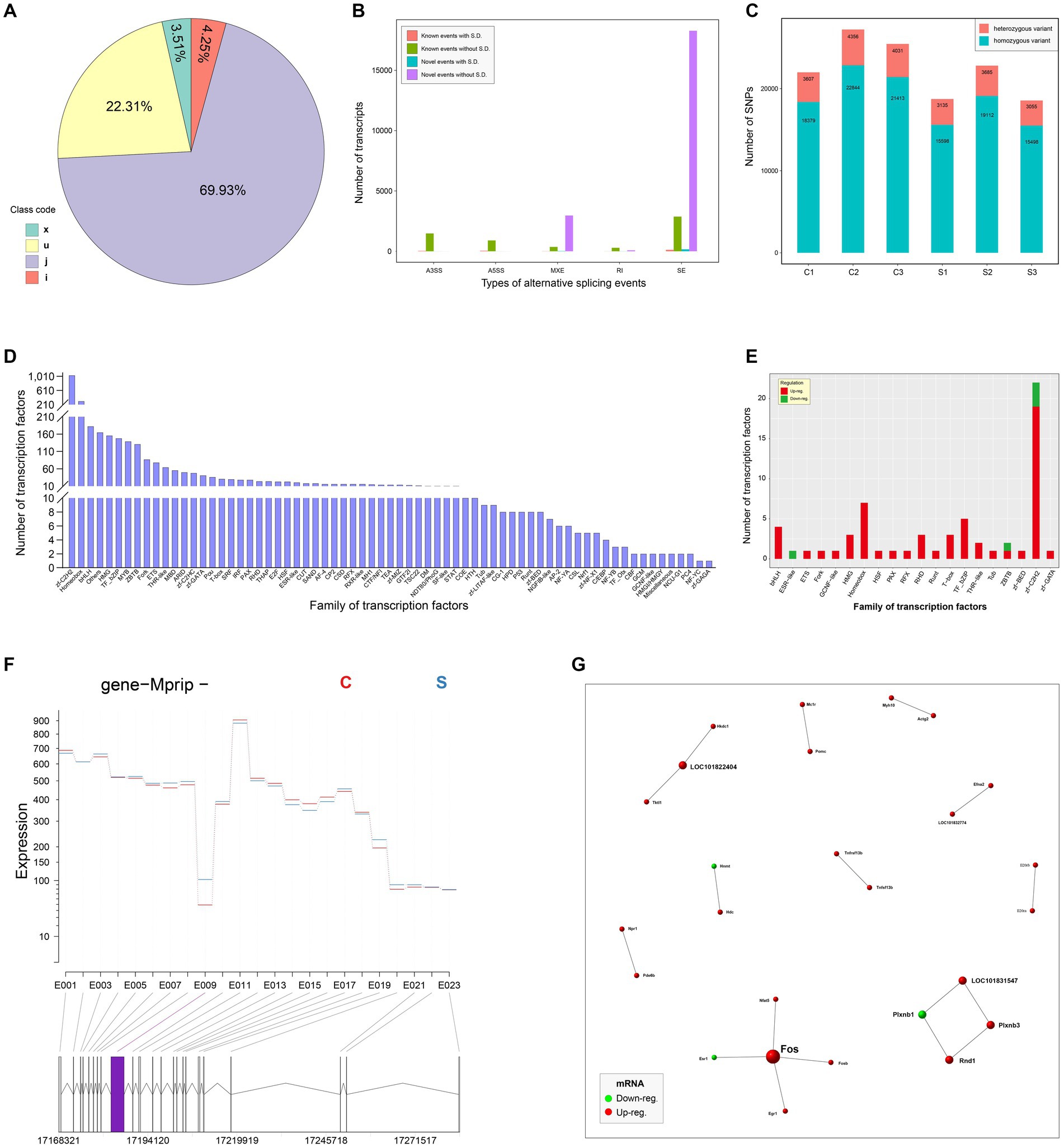
StringTie se usó para ensamblar las lecturas asignadas. Los resultados del ensamblaje se compararon con las transcripciones conocidas para obtener transcripciones sin anotaciones. Las transcripciones de los códigos de clase j, i, u y x, consideradas como nuevas transcripciones, fueron anotadas funcionalmente y listadas en el Archivo Suplementario 16. La proporción de cada código de clase se muestra en la Figura 5A. Se analizaron cinco tipos de eventos de empalme alternativos mediante el software rMATS (v3.2.5). El SE y el RI exhibieron la mayor y la menor cantidad de eventos de empalme alternativos, respectivamente (Figura 5B). Los sitios SNP fueron analizados por el programa Varscan. El número de variantes heterocigóticas y homocigóticas se mostró en la Figura 5C. Los factores de transcripción y sus propias familias se predijeron a través de la comparación con los del AnimalTFDB. La figura 5D muestra el número de factores de transcripción en cada familia. De las familias identificadas, se demostró que 20 contenían componentes regulados al alza, regulados a la baja o ambos (Figura 5E). El paquete DEXSeq se utilizó para analizar los datos de RNA-seq para identificar el uso diferencial del exón, como se muestra en el archivo suplementario 17. La Figura 5F reveló un gen representativo con uso diferencial de exones. Los DEG se analizaron exhaustivamente en la base de datos STRING para revelar posibles PPI (Puntuación > 0,95) para construir una red PPI, que incluye 26 nodos y 17 bordes (Figura 5G). Los nodos rojos y verdes indicaron genes regulados al alza y a la baja, respectivamente.
3.8 Validación de la expresión génica mediante RT-qPCR
Se seleccionaron tres DEG regulados al alza y uno a la baja para validar el perfil de expresión génica a través de RT-qPCR. Los tres genes regulados al alza incluían el genoma SVA, Nfkbia y Phlda2 (Archivo suplementario 12); el gen regulado a la baja fue Txnip (Ficha suplementaria 13). La detección de RT-qPCR demostró que la tendencia de expresión de los DEG era consistente con el resultado del análisis de RNA-seq (Figura 6). Debido a que el grupo C no inoculó SVA, la diferenciación de la expresión del genoma de SVA fue extremadamente significativa entre ambos grupos (Figura 6, Izquierda superior). No hubo necesidad de un análisis estadístico al respecto.
4 Discusión
Picornaviridae es una familia bien caracterizada dentro de los virus de ARN de cadena plus. El SVA es un picornavirus típico. Su genoma es solo un ARNm monocatenario de sentido positivo, que alberga una cola de poli(A) de 3′ pero no una estructura de tapa de 5′. En otras palabras, un virión SVA tiene un solo ARNm, que sin embargo no es la transcripción viral. Los picornavirus, aunque estructuralmente simples, posiblemente tienen efectos significativos sobre las funciones fisiológicas de sus huéspedes. Después de la entrada del virión en una célula, se liberará un genoma de picornavirus en el citosol. Este genoma se basa en la maquinaria de traducción del huésped para iniciar la traducción de la poliproteína, o sirve como plantilla para sintetizar un antigenoma, que se utilizará como plantilla para sintetizar otro genoma. El genoma naciente se puede utilizar como plantilla para la siguiente ronda de traducción o replicación, y alternativamente se empaqueta en un virión (43). Por lo tanto, aunque el SVA como tal no tiene el concepto de transcriptoma viral, su infección puede ejercer un impacto significativo en el transcriptoma celular. Esto nos llevó a realizar el presente estudio para descubrir el cambio transcriptómico en las células infectadas por SVA.
Un SVA competente para la replicación fue rescatado previamente de su clon de ADNc en nuestro laboratorio (27). El SVA de pasaje 5 se utilizó aquí como virus modelo. A pesar de la inoculación de SVA con MOI de 2,5, tres monocapas celulares no mostraron CPE visible a 12 hpi (Figura 1). Debido a que demostramos previamente que la infección por SVA condujo a cambios celulares significativos tanto en los perfiles proteómicos como en los metabolómicos a los 12 hpi, se podría postular que el transcriptoma celular también se vería afectado a los 12 hpi hasta cierto punto. El RNA-seq reconoció un total de 20.374 genes en los seis grupos, pero que contenían más de 3.000 genes con un valor de FPKM = 0. La correlación de la expresión génica es un indicador importante para demostrar la fiabilidad del experimento y la razonabilidad de las muestras. Un cierto coeficiente de correlación, si está entre 0,8 y 1,0, indicaría la correlación extremadamente fuerte entre dos grupos. El análisis de correlación actual mostró una correlación intragrupo extremadamente fuerte, pero una correlación intergrupal débil (Figura 2D), lo que implica que los datos de RNA-seq eran confiables.
Los datos de RNA-seq se sometieron a un análisis adicional sobre la expresión de diferenciación. El resultado reconoció en total 565 DEG regulados al alza y 63 regulados a la baja. De estos DEGs, se seleccionaron cinco genes representativos (tres genes regulados al alza y dos regulados a la baja) para el análisis de RT-qPCR con el fin de validar preliminarmente el perfil de los DEGs. Excepto el gen DEG, Tcta regulado a la baja (datos no mostrados), los otros cuatro mostraron sus tendencias de expresión consistentes con el resultado del análisis de RNA-seq (Figura 6). En nuestro estudio anterior sobre proteómica comparativa entre células infectadas y no infectadas por SVA, identificamos un total de 305 DEP (proteínas expresadas diferencialmente) reguladas al alza y 56 reguladas a la baja (25). Independientemente del estudio actual o del anterior, el número de componentes regulados al alza fue mucho mayor que el de los regulados a la baja. Tal resultado fue consistente con nuestra postulación de que los DEG compartían una tendencia de regulación similar con los DEP entre los grupos infectados y no infectados por SVA. De los DEGs identificados en el grupo S, el genoma SVA fue estadísticamente más significativo en el nivel de expresión (Ficha suplementaria 12). Debido al grupo C sin inoculación de SVA, tanto los análisis de enriquecimiento de GO como los de KEGG excluyeron los datos del genoma de SVA, y otros con infinito positivo o negativo (Archivos suplementarios 12, 13). El análisis de enriquecimiento de KEGG mostró que varios DEG estaban significativamente enriquecidos en muchas vías relacionadas con la inmunidad, como la vía de señalización del TNF, la vía de señalización de la IL-17, la vía de señalización del receptor tipo Toll y la vía de señalización del receptor de células B (Figura 4F). El análisis de enriquecimiento de GO también reveló algunos términos estadísticamente significativos asociados con las respuestas inmunitarias, por ejemplo, la respuesta a la interleucina-1 y la respuesta celular al factor de necrosis tumoral (Figura 4A). Al igual que la conclusión a la que se llegó en un informe anterior (44), los resultados actuales también sugieren que la infección por SVA puede ser capaz de inducir respuestas inmunitarias significativas, especialmente la respuesta inmunitaria innata, en huéspedes en una fase temprana de la infección.
Además, los datos de RNA-seq se sometieron a análisis en profundidad, relativos a SNP, factores de transcripción, IBP, etc. El análisis de los eventos de SNP indicó que no hubo una diferenciación significativa en el número de eventos de SNP entre los grupos C y S (Figura 5C), lo que implica que la infección por SVA no tenía la capacidad de inducir la aparición de eventos de SNP en el genoma del huésped. Vale la pena señalar que de las 20 familias de factores de transcripción estadísticamente significativas, la mayoría de ellas solo contienen componentes regulados al alza (Figura 5E). Este resultado implica que la infección por SVA puede estimular notablemente múltiples vías transcripcionales, lo que da lugar a DEG regulados al alza mucho más que a DEG regulados a la baja. La base de datos STRING se utilizó aquí para desentrañar los supuestos IBP en células infectadas por SVA. El resultado reveló que no se había formado una red de interacción complicada entre 26 DEP putativos (Figura 5G). Aunque los 26 DEPs no incluyen ninguna proteína relacionada con SVA, no se puede descartar la posibilidad de que las proteínas relacionadas con SVA interactúen con proteínas celulares, ya que la información de las proteínas SVA no se ha depositado en la base de datos STRING.
En los últimos 20 años ha surgido SVA en muchos países y regiones. Todavía se ha considerado como un virus emergente. La selección natural ha sido una fuerza evolutiva primaria que ha afectado el sesgo de uso de codones SVA (45). El análisis multiómico proporciona un enfoque integrado para facilitar estudios en profundidad sobre la virología, especialmente sobre la interacción de los virus con sus huéspedes. Sobre la base de nuestras investigaciones previas sobre proteómica y metabolómica, se demostró que la infección por SVA podría conducir a cambios significativos en los componentes intrínsecos celulares incluso en una etapa temprana de la infección (25, 26). Con el fin de analizar comparativamente los perfiles transcritómicos entre células infectadas y no infectadas por SVA, realizamos el presente estudio. En resumen, los resultados actuales revelaron que la mayoría de los DEG eran genes regulados al alza, lo que indica que la infección por SVA estimuló positivamente el inicio de la transcripción en las células. Los análisis de enriquecimiento de GO y KEGG demostraron que el SVA podría afectar marcadamente a las vías relacionadas con la inmunidad en las células, mientras que el mecanismo aún no se había dilucidado.
Declaración de disponibilidad de datos
Los datos presentados en el estudio están depositados en el repositorio del NCBI, número de acceso PRJNA1100277.
Contribuciones de los autores
YL: Redacción – borrador original, Metodología, Análisis formal. HC: Redacción: borrador original, metodología, investigación, curación de datos. YJ: Escritura – revisión y edición, análisis formal. ZL: Escritura – revisión y edición, Investigación. JW: Escritura – revisión y edición, Software. FL: Redacción – revisión y edición, Redacción – borrador original, Supervisión, Conceptualización.
Financiación
El/los autor/es declaran/n que se recibió apoyo financiero para la investigación, autoría y/o publicación de este artículo. Este trabajo fue apoyado por la Fundación Nacional de Ciencias Naturales de China (Subvención No. 32273000), el Proyecto de Demostración de Qingdao para el Beneficio de las Personas de la Ciencia y las Técnicas (Subvención No. 24-2-8-xdny-4-nsh) y el Programa de Innovación de Postgrado de la Universidad Agrícola de Qingdao (QNYCX24043).
Reconocimientos
Agradecemos a Shanghai Bioprofile Biotechnology Co., Ltd. (Shanghai, China) por brindar asistencia técnica en RNA-seq. También agradecemos a Xianggan Cui por su ayuda en el soporte técnico.
Conflicto de intereses
YJ fue empleado de Qingdao Zhongren-OLand Bioengineering Co., Ltd.
El resto de los autores declaran que la investigación se llevó a cabo en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un potencial conflicto de intereses.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o afirmación que pueda ser hecha por su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fvets.2024.1431879/full#supplementary-material
Referencias
1. Pasma, T, Davidson, S, y Shaw, SL. Enfermedad vesicular idiopática en cerdos en Manitoba. ¿Puede Vet J. (2008) 49:84–5.
2. Singh, K, Corner, S, Clark, S, Scherba, G, y Fredrickson, R. Virus del Valle de Séneca y lesiones vesiculares en un cerdo con enfermedad vesicular idiopática. J. Vet. Sci. Technol. (2012) 3:1–3. doi: 10.4172/2157-7579.1000123
3. Leme, RA, Zotti, E, Alcantara, BK, Oliveira, MV, Freitas, LA, Alfieri, AF, et al. Senecavirus a: una infección vesicular emergente en piaras de cerdos brasileños. Transbound Emerg Dis. (2015) 62:603–11. doi: 10.1111/tbed.12430
Resumen de PubMed | Texto completo de Crossref | Google Académico
4. Sun, D, Vannucci, F, Knutson, TP, Corzo, C, y Marthaler, DG. Emergencia y secuencia del genoma completo de Senecavirus a en Colombia. Transbound Emerg Dis. (2017) 64:1346–9. doi: 10.1111/tbed.12669
Resumen de PubMed | Texto completo de Crossref | Google Académico
5. Wu, Q, Zhao, X, Bai, Y, Sun, B, Xie, Q y Ma, J. La primera identificación y genoma completo de Senecavirus, un cerdo afectado con enfermedad vesicular idiopática en China. Transbound Emerg Dis. (2017) 64:1633–40. doi: 10.1111/tbed.12557
6. Leme, RA, Alfieri, AF, y Alfieri, AA. Actualización sobre la infección por Senecavirus en cerdos. Virus. (2017) 9:170. doi: 10.3390/v9070170
Resumen de PubMed | Texto completo de Crossref | Google Académico
7. Leme, RA, Oliveira, TE, Alcántara, BK, Headley, SA, Alfieri, AF, Yang, M, et al. Manifestaciones clínicas de la infección por Senecavirus a en cerdos neonatos, Brasil, 2015. Emerg Infect Dis. (2016) 22:1238–41. doi: 10.3201/eid2207.151583
Resumen de PubMed | Texto completo de Crossref | Google Académico
8. Fernandes, MHV, de Lima, M, Joshi, LR, y Diel, DG. Un clon infeccioso virulento y patógeno de Senecavirus a. J Gen Virol. (2021) 102:001643. doi: 10.1099/jgv.0.001643
Resumen de PubMed | Texto completo de Crossref | Google Académico
9. Oliveira, TES, Leme, RA, Agnol, AMD, Gerez, JR, Pelaquim, IF, Miyabe, FM, et al. El virus del Valle de Séneca induce inmunodepresión en lechones lactantes por apoptosis selectiva de linfocitos B. Microb Pathog. (2021) 158:105022. doi: 10.1016/j.micpath.2021.105022
Resumen de PubMed | Texto completo de Crossref | Google Académico
10. Hellen, CU, y de Breyne, S. Un grupo distinto de sitios de entrada ribosómica interna similares a hepacivirus/pestivirus en miembros de diversos géneros de picornavirus: evidencia de intercambio modular de elementos de ARN funcionales no codificantes por recombinación. J Virol. (2007) 81:5850–63. doi: 10.1128/JVI.02403-06
Resumen de PubMed | Texto completo de Crossref | Google Académico
11. Willcocks, MM, Locker, N, Gomwalk, Z, Royall, E, Bakhshesh, M, Belsham, GJ, et al. Características estructurales del elemento del sitio de entrada del ribosoma interno (IRES) del virus Seneca Valley: un picornavirus con un IRES similar a un pestivirus. J Virol. (2011) 85:4452–61. doi: 10.1128/JVI.01107-10
Resumen de PubMed | Texto completo de Crossref | Google Académico
12. Zhao, D, Li, Y, Li, Z, Zhu, L, Sang, Y, Zhang, H, et al. Sólo catorce poli(a)s de 3′ extremos son suficientes para rescatar a Senecavirus a de su clon de ADNc, pero inadecuados para cumplir con los requisitos de replicación viral. Virus Res. (2023) 328:199076. doi: 10.1016/j.virusres.2023.199076
Resumen de PubMed | Texto completo de Crossref | Google Académico
13. Liu, F, Wang, Q, Huang, Y, Wang, N y Shan, H. Una revisión de 5 años del Senecavirus a en China desde su aparición en 2015. Front Vet Sci. (2020) 7:567792. doi: 10.3389/fvets.2020.567792
Resumen de PubMed | Texto completo de Crossref | Google Académico
14. Choudhury, SM, Ma, X, Zeng, Z, Luo, Z, Li, Y, Nian, X, et al. Senecavirus a 3D interactúa con NLRP3 para inducir la producción de IL-1β mediante la activación de las señales NF-κB y de canal iónico. Microbiol Spectr. (2022) 10:E0209721. doi: 10.1128/spectrum.02097-21
Resumen de PubMed | Texto completo de Crossref | Google Académico
15. Liu, H, Li, K, Chen, W, Yang, F, Cao, W, Zhang, K, et al. Senecavirus, una proteína 2B, suprime la producción de interferón tipo I al inducir la degradación de MAVS. Mol Immunol. (2022) 142:11–21. doi: 10.1016/j.molimm.2021.12.015
Resumen de PubMed | Texto completo de Crossref | Google Académico
16. Zhao, K, Guo, XR, Liu, SF, Liu, XN, Han, Y, Wang, LL, et al. Las proteínas 2B y 3C de Senecavirus a antagonizan la actividad antiviral de DDX21 a través de la degradación dependiente de caspasas de DDX21. Frente Immunol. (2022) 13:951984. doi: 10.3389/fimmu.2022.951984
Resumen de PubMed | Texto completo de Crossref | Google Académico
17. Duhan, L, Kumari, D, Naime, M, Parmar, VS, Chhillar, AK, Dangi, M, et al. Transcriptómica de una sola célula: antecedentes, tecnologías, aplicaciones y desafíos. Mol Biol Rep. (2024) 51:600. doi: 10.1007/s11033-024-09553-y
Resumen de PubMed | Texto completo de Crossref | Google Académico
18. Maher, CA, Kumar-Sinha, C, Cao, X, Kalyana-Sundaram, S, Han, B, Jing, X, et al. Secuenciación del transcriptoma para detectar fusiones génicas en cáncer. Naturaleza. (2009) 458:97–101. doi: 10.1038/nature07638
Resumen de PubMed | Texto completo de Crossref | Google Académico
19. Zeng, J, Cao, D, Yang, S, Jaijyan, DK, Liu, X, Wu, S, et al. Perspectivas sobre el transcriptoma del citomegalovirus humano: una revisión exhaustiva. Virus. (2023) 15:1703. doi: 10.3390/v15081703
Resumen de PubMed | Texto completo de Crossref | Google Académico
20. Deb, R, Sengar, GS, Sonowal, J, Pegu, SR, Das, PJ, Singh, I, et al. Las firmas del transcriptoma del tejido huésped infectado con el virus de la peste porcina africana revelan una expresión diferencial de oncogenes asociados. Arch Virol. (2024) 169:54. doi: 10.1007/s00705-023-05959-4
21. Wen, W, Yin, M, Zhang, H, Liu, T, Chen, H, Qian, P, et al. Seneca Valley virus 2C y 3C inhiben la producción de interferón tipo I al inducir la degradación de RIG-I. Virología. (2019) 535:122–9. doi: 10.1016/j.virol.2019.06.017
Resumen de PubMed | Texto completo de Crossref | Google Académico
22. Hou, L, Dong, J, Zhu, S, Yuan, F, Wei, L, Wang, J, et al. El virus del valle de Séneca activa la autofagia a través de las vías PERK y ATF6 UPR. Virología. (2019) 537:254–63. doi: 10.1016/j.virol.2019.08.029
Resumen de PubMed | Texto completo de Crossref | Google Académico
23. Li, L, Bai, J, Fan, H, Yan, J, Li, S y Jiang, P. La enzima conjugadora de ubiquitina E2 UBE2L6 promueve la proliferación de Senecavirus a al estabilizar la ARN polimerasa viral. Ruta PLoS. (2020) 16:e1008970. doi: 10.1371/journal.ppat.1008970
Resumen de PubMed | Texto completo de Crossref | Google Académico
24. Liu, T, Li, X, Wu, M, Qin, L, Chen, H y Qian, P. Seneca Valley virus 2C y 3C(pro) inducen la apoptosis a través de la vía intrínseca mediada por mitocondrias. Microbiol frontal. (2019) 10:1202. doi: 10.3389/fmicb.2019.01202
Resumen de PubMed | Texto completo de Crossref | Google Académico
25. Liu, F, Ni, B y Wei, R. Perfil proteómico comparativo: los metabolismos celulares se ven afectados principalmente en las células inoculadas con Senecavirus A en una etapa temprana de la infección. Virus. (2021) 13:1036. doi: 10.3390/v13061036
Resumen de PubMed | Texto completo de Crossref | Google Académico
26. Liu, F, Ni, B y Wei, R. Células no infectadas y del Senecavirus A en la etapa temprana de la infección: perfiles metabolómicos comparativos. Las células frontales infectan el microbiol. (2022) 11:736506. doi: 10.3389/fcimb.2021.736506
Resumen de PubMed | Texto completo de Crossref | Google Académico
27. Liu, F, Huang, Y, Wang, Q, Li, J y Shan, H. Rescate de Senecavirus a para descubrir perfiles de mutación de sus progenies durante 80 pasajes seriados in vitro. Microbiol veterinario. (2021) 253:108969. doi: 10.1016/j.vetmic.2020.108969
Resumen de PubMed | Texto completo de Crossref | Google Académico
28. Zhao, X, Wu, Q, Bai, Y, Chen, G, Zhou, L, Wu, Z, et al. Análisis filogenético y genómico de siete aislamientos de senecavirus a en China. Transbound Emerg Dis. (2017) 64:2075–82. doi: 10.1111/tbed.12619
Resumen de PubMed | Texto completo de Crossref | Google Académico
29. Ding, L, Li, Q, Chakrabarti, J, Muñoz, A, Faure-Kumar, E, Ocadiz-Ruiz, R, et al. MiR130b de Schlafen4(+) MDSCs estimula la proliferación epitelial y se correlaciona con cambios preneoplásicos previos al cáncer gástrico. Tripa. (2020) 69:1750–61. doi: 10.1136/gutjnl-2019-318817
Resumen de PubMed | Texto completo de Crossref | Google Académico
30. Martin, M. Cutadapt elimina las secuencias de adaptador de las lecturas de secuenciación de alto rendimiento. EMBnet J. (2011) 17:10–2. doi: 10.14806/ej.17.1.200
31. Kim, D, Paggi, JM, Park, C, Bennett, C, y Salzberg, SL. Alineación y genotipado del genoma basado en grafos con HISAT2 y el genotipo HISAT. Nat Biotechnol. (2019) 37:907–15. doi: 10.1038/s41587-019-0201-4
Resumen de PubMed | Texto completo de Crossref | Google Académico
32. Qiao, J, Li, H y Li, Y. La suplementación dietética con Clostridium butyricum modifica significativamente el perfil transcriptómico hepático en lechones destetados. J Anim Physiol Anim Nutr. (2020) 104:1410–23. doi: 10.1111/jpn.13326
Resumen de PubMed | Texto completo de Crossref | Google Académico
33. Anders, S, Pyl, PT y Huber, W. HTSeq: un marco de Python para trabajar con datos de secuenciación de alto rendimiento. Bioinformática (2015) 31:166–169. doi: 10.1093/bioinformatics/btu638, HTSeq: un marco de Python para trabajar con datos de secuenciación de alto rendimiento
Resumen de PubMed | Texto completo de Crossref | Google Académico
34. Li, D, Zand, MS, Dye, TD, Goniewicz, ML, Rahman, I, y Xie, Z. Una evaluación de los métodos de análisis diferencial de RNA-seq. PLoS Uno. (2022) 17:e0264246. doi: 10.1371/journal.pone.0264246
Resumen de PubMed | Texto completo de Crossref | Google Académico
35. Yu, G, Wang, LG, Han, Y y He, QY. clusterProfiler: un paquete de R para comparar temas biológicos entre grupos de genes. OMICS. (2012) 16:284–7. doi: 10.1089/omi.2011.0118
Resumen de PubMed | Texto completo de Crossref | Google Académico
36. Kovaka, S, Zimin, AV, Pertea, GM, Razaghi, R, Salzberg, SL y Pertea, M. Ensamblaje del transcriptoma a partir de alineaciones de secuenciación de ARN de lectura larga con StringTie2. Genoma Biol. (2019) 20:278. doi: 10.1186/s13059-019-1910-1
Resumen de PubMed | Texto completo de Crossref | Google Académico
37. Shen, S, Park, JW, Lu, ZX, Lin, L, Henry, MD, Wu, YN, et al. rMATS: detección robusta y flexible de empalme alternativo diferencial a partir de datos replicados de RNA-Seq. Proc Natl Acad Sci USA. (2014) 111:E5593–601. doi: 10.1073/pnas.1419161111
Resumen de PubMed | Texto completo de Crossref | Google Académico
38. Koboldt, DC, Chen, K, Wylie, T, Larson, DE, McLellan, MD, Mardis, ER, et al. VarScan: detección de variantes en secuenciación masiva paralela de muestras individuales y agrupadas. Bioinformática. (2009) 25:2283–5. doi: 10.1093/bioinformatics/btp373
Resumen de PubMed | Texto completo de Crossref | Google Académico
39. Zhang, HM, Chen, H, Liu, W, Liu, H, Gong, J, Wang, H, et al. AnimalTFDB: una base de datos completa de factores de transcripción de animales. Ácidos nucleicos Res. (2012) 40:D144-9. doi: 10.1093/nar/gkr965
Resumen de PubMed | Texto completo de Crossref | Google Académico
40. Anders, S, Reyes, A y Huber, W. Detección del uso diferencial de exones a partir de datos de RNA-seq. Genoma Res. (2012) 22:2008–17. doi: 10.1101/gr.133744.111
Resumen de PubMed | Texto completo de Crossref | Google Académico
41. Szklarczyk, D, Morris, JH, Cook, H, Kuhn, M, Wyder, S, Simonovic, M, et al. La base de datos STRING en 2017: redes de asociación proteína-proteína de calidad controlada, ampliamente accesibles. Ácidos nucleicos Res. (2017) 45:D362-8. doi: 10.1093/nar/gkw937
Resumen de PubMed | Texto completo de Crossref | Google Académico
42. Livak, KJ, y Schmittgen, TD. Análisis de datos relativos de expresión génica mediante PCR cuantitativa en tiempo real y el método 2(-Delta Delta C(T)). Métodos. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
43. Kinobe, R, Wiyatno, A, y Artika, IM. Información sobre el enterovirus A71: una revisión. Rev Med Virología. (2022) 32:E2361. doi: 10.1002/rmv.2361
44. Wang, J, Mou, C, Wang, M, Pan, S y Chen, Z. Análisis del transcriptoma de las células infectadas por el seneca A: el interferón tipo I es un factor antiviral crítico. Microb Pathog. (2020) 147:104432. doi: 10.1016/j.micpath.2020.104432
Resumen de PubMed | Texto completo de Crossref | Google Académico
45. Zeng, W, Yan, Q, Du, P, Yuan, Z, Sun, Y, Liu, X, et al. Dinámica evolutiva y análisis adaptativo del virus Seneca Valley. Infectar Genet Evol. (2023) 113:105488. doi: 10.1016/j.meegid.2023.105488
Resumen de PubMed | Texto completo de Crossref | Google Académico
Palabras clave: Senecavirus A, RNA-seq, transcriptómica, gen expresado diferencialmente, análisis de enriquecimiento, inmunidad, vía
Cita: Li Y, Chu H, Jiang Y, Li Z, Wang J y Liu F (2024) Análisis transcriptómico comparativo en células infectadas y no infectadas por Senecavirus A. Frente. Vet. Sci. 11:1431879. doi: 10.3389/fvets.2024.1431879
Editado por:
Mengmeng Zhao, Universidad de Foshan, China
Revisado por:
Pan Hu, Universidad de Jilin, China
Jianfa Wang, Universidad Agrícola de Heilongjiang Bayi, China
Derechos de autor © 2024 Li, Chu, Jiang, Li, Wang y Liu. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia Creative Commons Attribution License (CC BY).
*Correspondencia: Fuxiao Liu, laudawn@126.com
†Estos autores han contribuido igualmente a este trabajo
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente a las de sus organizaciones afiliadas, o las del editor, de los editores y de los revisores. Cualquier producto que puede ser evaluada en este artículo o afirmación que puede ser hecha por su El fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía