
Caracterización del proteoma plasmático de perros adultos sanos

Caracterización del proteoma plasmático de perros adultos sanos
 Pavlos G. Doulidis1
Pavlos G. Doulidis1 Benno Kuropka2
Benno Kuropka2 Carolina Frizzo Ramos3,4
Carolina Frizzo Ramos3,4 Alexandro Rodríguez-Rojas1*
Alexandro Rodríguez-Rojas1* Iwan A. Burgener1*
Iwan A. Burgener1*- 1División de Medicina Interna de Pequeños Animales, Departamento de Pequeños Animales y Caballos, Universidad de Medicina Veterinaria de Viena, Viena, Austria
- número arábigoInstituto de Química y Bioquímica, Freie Universität Berlin, Berlín, Alemania
- 3Instituto Interuniversitario de Investigación Messerli, Universidad de Medicina de Viena, Viena, Austria
- 4Centro Clínico para Pequeños Animales, Universidad de Medicina Veterinaria de Viena, Viena, Austria
Introducción: Los análisis de sangre son una herramienta diagnóstica ampliamente utilizada en medicina veterinaria, ya que el diagnóstico y las intervenciones terapéuticas a menudo se basan en biomarcadores sanguíneos. Sin embargo, los biomarcadores disponibles en medicina veterinaria a menudo carecen de sensibilidad o especificidad. La tecnología proteómica basada en espectrometría de masas se ha utilizado ampliamente en el análisis de fluidos biológicos. Ofrece un excelente potencial para una caracterización más completa del proteoma plasmático en medicina veterinaria.
Métodos: En este estudio, nuestro objetivo fue identificar y cuantificar las proteínas plasmáticas en una cohorte de perros sanos y comparar dos técnicas para agotar las proteínas plasmáticas de alta abundancia para permitir la detección de proteínas de menor abundancia mediante cuantificación sin marcadores, cromatografía líquida y espectrometría de masas. Utilizamos plasma excedente de litio-heparina de 30 perros sanos, subdivididos en cinco grupos de plasma agrupado de 6 individuos seleccionados al azar cada uno. En primer lugar, utilizamos un kit comercial para agotar las proteínas plasmáticas de alta abundancia. En segundo lugar, empleamos un método interno para eliminar la albúmina utilizando Blue-Sepharose.
Resultados y discusión: Entre todas las muestras, algunas de las proteínas más abundantes identificadas fueron la apolipoproteína A y B, la albúmina, la alfa-2-macroglobulina, la cadena beta del fibrinógeno, la fibronectina, el complemento C3, la serotransferrina y el factor V de la coagulación. Sin embargo, ninguna de las técnicas de depleción logró un agotamiento significativo de proteínas altamente abundantes. A pesar de esta limitación, pudimos detectar y cuantificar muchas proteínas clínicamente relevantes. La determinación del proteoma canino sano es un primer paso crucial para establecer un proteoma de referencia para el plasma canino. Tras el enriquecimiento, este proteoma de referencia puede utilizarse posteriormente para identificar marcadores proteicos asociados a diferentes enfermedades, contribuyendo así al diagnóstico y pronóstico de diversas patologías.
Introducción
Los biomarcadores se definen como rasgos que pueden medirse como indicadores de un proceso patogénico o de una respuesta farmacológica al tratamiento y que pueden evaluarse de forma objetiva (1). Los ensayos enzimáticos e inmunoensayos son los métodos más utilizados para cuantificar biomarcadores altamente abundantes. Sin embargo, se ha informado de que la tasa de introducción de nuevos biomarcadores en medicina es inferior a dos por año (2), y la adopción de estos biomarcadores en medicina veterinaria a menudo se retrasa.
La proteómica basada en la espectrometría de masas (MS) se ha convertido en una tecnología poderosa en la investigación biológica (3-5) y médica (6, 7). Ofrece la capacidad de caracterizar de forma exhaustiva el proteoma plasmático, contribuyendo así al descubrimiento de nuevos biomarcadores (8-10). A diferencia de las técnicas tradicionales, la espectrometría de masas de péptidos de alta precisión y espectros de fragmentación derivados de la digestión de proteínas de interés específicas de la secuencia (10). La proteómica es altamente específica debido a la singularidad de las masas y secuencias peptídicas, en contraste con las pruebas enzimáticas colorimétricas y los inmunoensayos (11).
Entre varios enfoques, la cromatografía líquida-espectrometría de masas en tándem (LC-MS/MS) es a menudo un método preferido en la investigación médica debido a su alta especificidad analítica y sensibilidad, lo que permite la detección y cuantificación de proteínas de baja abundancia, así como fármacos y metabolitos (12). En consecuencia, las técnicas proteómicas basadas en la EM están ganando cada vez más interés en la medicina veterinaria de pequeños animales. Aunque la investigación sobre el análisis proteómico basado en la EM en medicina veterinaria no es tan extensa como en la investigación clínica en humanos, se han reportado estudios basados en la EM que analizan fluidos biológicos de perros con diversas enfermedades (13-19). Dada la escasez de datos técnicos y médicos en la literatura veterinaria, el escaso número de proteínas detectadas en el suero canino en los estudios mencionados anteriormente y la importancia del análisis proteómico para la detección de nuevos biomarcadores o redes de biomarcadores en el plasma canino, es necesario explorar nuevos métodos para estudiar los biomarcadores sanguíneos.
El plasma se refiere a la porción líquida de sangre no coagulada que queda después de eliminar todos los tipos de células. La heparina es uno de los anticoagulantes más utilizados para la preparación plasmática, actuando a través de la activación antitrombina. Cuando se desencadena la formación de coágulos en el plasma para formar suero, el fibrinógeno y otros factores de coagulación se agotan, mientras que se liberan péptidos como el fibrinopéptido A y B (20-22). En las últimas décadas, el número de proteínas identificadas en el plasma humano ha aumentado exponencialmente (23-25). El análisis proteómico del plasma ofrece la ventaja de cuantificar proteínas de alta abundancia, como el fibrinógeno y, parcialmente, otros factores de coagulación que ya no están presentes en el suero, lo que podría revelar patrones específicos de enfermedades. Sin embargo, debido al rango dinámico altamente alto del plasma, la identificación de proteínas de menor abundancia mediante LC-MS puede ser un desafío. Técnicas como la depleción de anticuerpos de proteínas plasmáticas abundantes como la albúmina y el fibrinógeno, así como el fraccionamiento plasmático extensivo, se han utilizado con éxito para facilitar la detección de proteínas de baja abundancia en muestras humanas (26-28). Estas técnicas también se han combinado con éxito para identificar varios miles de proteínas (29-31).
La electroforesis bidimensional en gel (2-DE) se ha utilizado ampliamente para el análisis proteómico (32, 33). Sin embargo, este método tiene limitaciones para identificar eficazmente proteínas de baja abundancia, y su rango dinámico es limitado. Las metodologías sin gel han ganado atención en los últimos años, ya que permiten la determinación y cuantificación de una gama más amplia de proteínas (34). La necesidad de detectar nuevos biomarcadores pronósticos para predecir los resultados de las enfermedades ha llevado a que la investigación proteómica en medicina canina se centre en las enfermedades infecciosas, siendo la leishmaniosis el ejemplo más destacado (14, 35, 36), seguida de enfermedades como la babesiosis (13, 37-39), la ehrlichiosis (40) y la infección por parvovirus (17). Algunos estudios en pacientes con leishmaniosis y babesiosis encontraron una disminución significativa de la apolipoproteína A, lo que puede reducir la capacidad del individuo para responder al daño oxidativo. Más allá del campo de las enfermedades infecciosas, el análisis proteómico ha proporcionado nuevos conocimientos sobre la nefrología veterinaria, revelando que un aumento en proteínas como la proteína de unión al retinol predice el daño renal antes de que se desarrolle azotemia (41-43), y en endocrinología veterinaria, descubriendo el papel de la apolipoproteína I en la obesidad canina (44, 45). Según la literatura, las muestras más analizadas en medicina de pequeños animales son el suero y la saliva (46). También se han realizado estudios con otros fluidos y tejidos biológicos como el líquido cefalorraquídeo, la bilis, el hígado, el líquido sinovial, el miocardio (47-51) y las muestras de heces (52). En la actualidad, no se dispone de un catálogo sistemático de proteínas plasmáticas caninas. Aunque el análisis proteómico del plasma canino se realiza menos que el análisis sérico, se han reportado algunos estudios sobre el plasma canino, con las proteínas plasmáticas abundantes diferenciales detectadas en perros con SIRS y MODS siendo 68, 12 en perros obesos con y sin disfunción metabólica relacionada con la obesidad y, finalmente, 87 en perros diagnosticados con síndrome de disfunción cognitiva canina. respectivamente (19, 53, 54).
Este estudio tiene como objetivo utilizar el análisis proteómico LC-MS de cuantificación sin marcadores para explorar la composición del plasma canino en perros sanos de diferentes razas. Estos hallazgos servirán como base para futuros estudios que involucren cohortes más grandes de enfermedades específicas, con el objetivo de detectar nuevos biomarcadores u obtener una mejor comprensión de las redes de biomarcadores en diversas enfermedades.
Materiales y métodos
Animales y recogida de muestras
Perros clínicamente sanos (N = 30) sin signos de enfermedad en los últimos 2 meses se presentaron en la División de Medicina Interna de Pequeños Animales (Universidad Veterinaria de Viena, Viena, Austria) durante 6 meses para su examen clínico y toma de muestras de sangre. Participaron en un estudio (Ref: BMBWF 20221-0.210.26) realizado por la División de Medicina Interna de Pequeños Animales y el Instituto Interuniversitario de Investigación Messerli (Universidad Veterinaria de Viena, Austria). Antes de la inscripción, se obtuvo el consentimiento informado por escrito de los propietarios. Se consideraron perros de diferentes razas, pesos corporales y sexos, con edades comprendidas entre 1 y 10 años, para el estudio de proteómica. Los criterios de inclusión incluyeron una evaluación integral, que incluyó una anamnesis detallada, un examen físico y una toma de muestras de sangre realizadas por dos autores (DP y CF). Se realizó un hemograma completo (CSC), un perfil bioquímico sérico y mediciones de electrolitos. Además, se registró la puntuación de la condición corporal (escala Nestlé Purina: que va de 1-muy delgado a 9-obesidad significativa) de cada perro. Se excluyeron del estudio los perros menores de 1 año, con un peso inferior a 5 kg, y aquellos con signos patológicos o administración reciente de medicamentos en los últimos 2 meses. Asimismo, no se incluyeron perros con alteraciones significativas en alguno de los parámetros sanguíneos. La aleatorización y la selección de grupos se realizaron utilizando el paquete R «tidyverse».
Preparación de muestras para el análisis del proteoma
Las muestras de sangre se recogieron utilizando tubos Vacuette de 2 mL con heparina de litio 13×75 tapa verde-anillo blanco PREMIUM (Greiner Bio-One GmbH, Bad Haller Str. 32, 4550, Austria). Después de la centrifugación a 2.000 × g durante 5 minutos, el plasma se separó y almacenó a -21 °C. Se utilizaron cinco microlitros de plasma de cada perro de la cohorte de 30 individuos para crear grupos aleatorios. Creamos tres grupos separados que consistieron en cinco grupos, cada uno de los cuales contenía plasma agrupado de seis individuos para un volumen final de 30 μL. Uno de los grupos optó por 14 procedimientos de depleción de proteínas más abundantes utilizando un kit comercial (Top14 Abundant Protein Depletion Mini Spin Columns, Thermo Scientifics, Alemania) que se utilizó siguiendo las instrucciones del fabricante. El segundo grupo consistió en un procedimiento interno de depleción de albúmina utilizando Blue-Sepharose CL6B (GE Health Care, Alemania). El procedimiento consistió en mezclar 30 μL de piscinas de plasma con 100 μL de Sepharose azul preequilibrado con tampón fosfato (PBS, 150 mM, pH 7,2), incubación durante 30 min con agitación suave, separación del sobrenadante por centrifugación (4.000 × g durante 10 min) y separación del plasma sobrante que se utilizó para el procedimiento proteómico. El tercer grupo consistió en piscinas de plasma sin ningún tratamiento de depleción que se utilizaron directamente para la proteómica.
Se transfirieron cinco microlitros de plasma por piscina a un tubo que contenía 20 μL de tampón desnaturalizante de urea (6 M de urea, 2 M de tiourea y 10 mM de HEPES, pH 8,0). Los enlaces disulfuro de las proteínas plasmáticas se redujeron mediante la adición de 1 μL de ditiotreitol (10 mM, concentración de stock) y se incubaron durante 30 min a temperatura ambiente. Posteriormente, las muestras se alquilaron añadiendo 1 μL de solución de yodoacetamida (55 mM, concentración de stock) y se incubaron a temperatura ambiente durante otros 30 min en la oscuridad. Las muestras se diluyeron con cuatro volúmenes de tampón de bicarbonato de amonio (40 mM) y se digirieron durante la noche a 37 °C añadiendo 1 μg de tripsina proteasa (Thermo Scientific, Estados Unidos). Para acidificar las muestras, se añadió acetonitrilo al 5% y ácido trifluoroacético (TFA; concentración final) al 0,3% y, posteriormente, las muestras se desalinizaron utilizando C18 StageTips con discos de extracción Empore™ C18 (55). Los péptidos eluidos de los StageTips se secaron mediante centrifugación al vacío.
Cromatografía líquida-análisis por espectrometría de masas
Los péptidos se reconstituyeron en 40 μL de una solución que contenía 0,05% de TFA y 4% de acetonitrilo. A continuación, se aplicaron 2 μL de cada muestra a un sistema de cromatografía nanolíquida capilar Ultimate 3.000 de fase reversa conectado a un espectrómetro de masas Q Exactive HF (Thermo Fisher Scientific). Las muestras se inyectaron y concentraron en una columna trampa PepMap100 C18 [3 μm, 100 Å, 75 μm de diámetro interior (i.d.) × 20 mm, nanoViper; Thermo Scientific] que se equilibró con un 0,05% de TFA en agua. Después de cambiar la columna de trampa en línea, se realizaron separaciones de LC en una columna capilar Acclaim PepMap100 C18 (2 μm, 100 Å, 75 μm i.d. × 500 mm, nanoViper, Thermo Scientific) a un caudal de eluyente de 300 nL/min. La fase móvil A consistía en un 0,1% (v/v) de ácido fórmico en agua, mientras que la fase móvil B contenía un 0,1% (v/v) de ácido fórmico y un 80% (v/v) de acetonitrilo en agua. La columna se preequilibró con un 5% de fase móvil B, seguida de un aumento al 44% de fase móvil B durante 35 min. Los espectros de masas se adquirieron en un modo dependiente de los datos, utilizando un solo escaneo de sondeo MS (m/z 300-1.650) con una resolución de 60.000, y escaneos MS/MS de los 15 iones precursores más intensos con una resolución de 15.000. El tiempo de exclusión dinámica se fijó en 20 s y el control automático de ganancia se fijó en 3 × 106 y 1 × 105 para las exploraciones MS y MS/MS, respectivamente.
Procesamiento de datos y cuantificación sin etiquetas
Los datos brutos de MS y MS/MS se analizaron utilizando el paquete de software MaxQuant (versión 2.0.3.0) con el motor de búsqueda de péptidos Andromeda implementado (56). Los datos se buscaron en el proteoma de referencia Canis lupus familiaris (ID: UP000002254; descargado de Uniprot.org el 17.10.2022; 43.621 secuencias) utilizando los parámetros predeterminados y habilitando las opciones de cuantificación sin etiquetas (LFQ) y coincidencia entre corridas. El filtrado de datos y el análisis estadístico se realizaron utilizando el software Perseus 1.6.14 (57). Solo se utilizaron para el análisis posterior las proteínas identificadas y cuantificadas con valores de intensidad de LFQ en al menos tres (de cinco) réplicas dentro de al menos uno de los tres grupos experimentales. Los valores faltantes se reemplazaron de una distribución normal (imputación) utilizando la configuración predeterminada (ancho 0.3, desplazamiento descendente 1.8). Las diferencias medias de log2 veces entre los grupos se calcularon en Perseo mediante la prueba t de Student. Las proteínas con un cambio de intensidad mínimo de 2 veces en comparación con el control (cambio log2 veces ≥1 o cambio log2 veces ≤ −1) y un valor q ≤0,05 (valor p ajustado) se consideraron significativamente abundantes.
Análisis estadístico
Todas las comparaciones estadísticas entre grupos se realizaron utilizando la prueba t de student implementada por la plataforma computacional Perseus (57). Los valores p ajustados después de FDR (valores q) se consideraron significativos para valores inferiores a 0,05.
Resultados y discusión
En nuestro estudio, utilizamos un método LC-MS de cuantificación sin marcadores para analizar el plasma canino. Se utilizó el plasma de 30 individuos sanos. La edad media de los 30 perros fue de 5,7 años (rango de 1,3 a 9,9 años), el peso medio fue de 18,4 kg (rango de 5 a 37,5 kg), mientras que 15 perros eran machos (50%) y 15 perros eran hembras (50%). Dieciséis (60%) perros fueron castrados y 14 (40%) estaban intactos. Todos los perros incluidos tuvieron una puntuación de condición corporal ideal de 5/9. De estos perros, 9 eran mestizos, 3 Pastores Australianos, 2 Golden Retrievers, 2 Pastores Alemanes, 2 Pulis y 12 otras razas estaban representadas por un perro cada una. Estas muestras se seleccionaron aleatoriamente, de acuerdo con los criterios de inclusión, para evitar preservar la heterogenicidad y posteriormente se agruparon en cinco grupos de seis individuos utilizando un script informático, como se describe en la sección de material y métodos. Evaluamos el contenido de proteínas plasmáticas y evaluamos dos métodos de depleción para proteínas de alta abundancia. El primer método implicó el uso de un kit comercial diseñado para agotar las 14 proteínas plasmáticas humanas más abundantes (denominado «kit»), mientras que el segundo método empleó un enfoque interno de bajo costo utilizando Blue-Sepharose. El propósito del método Blue-Sepharose era específicamente eliminar la albúmina (conocida como «Blue-Sepharose»). Para optimizar el proceso, incorporamos un conjunto de muestras clínicas obtenidas de individuos sanos con perfiles normales bien establecidos de parámetros sanguíneos rutinarios.
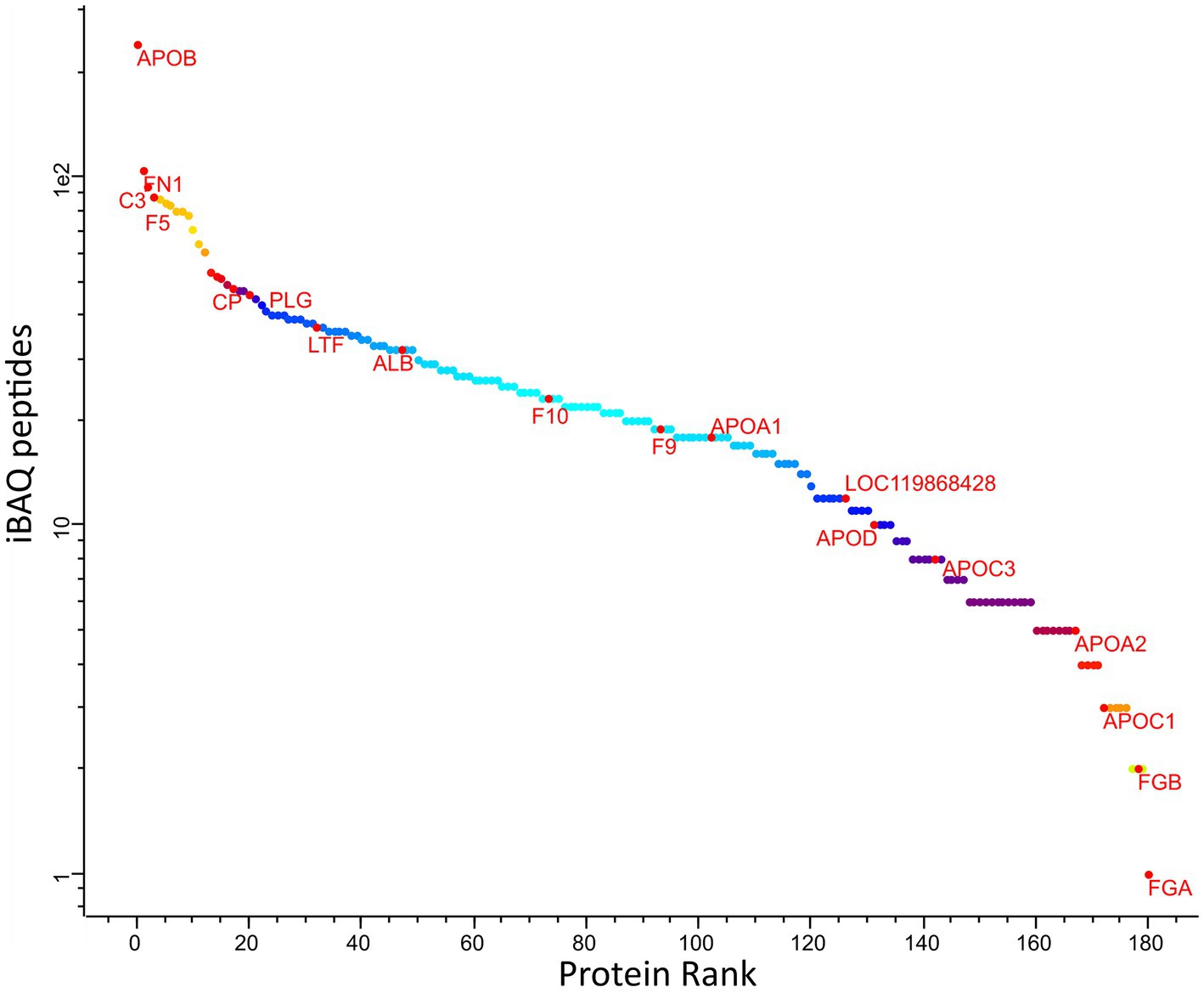
Todos los archivos de resultados de nuestros experimentos de EM se proporcionan en el material complementario. Inicialmente, identificamos un total de 282 proteínas. Posteriormente, se eliminaron las proteínas que no se identificaron en al menos 3 de las 5 réplicas de al menos uno de los tres grupos (Control, Kit, Blue-Sepharose). Este proceso de filtrado dio como resultado la cuantificación de 181 proteínas en las muestras de plasma. La Figura 1 ilustra los pasos de procesamiento de datos mencionados anteriormente. Para la descripción de las proteínas, se utilizaron principalmente las identificaciones de genes. En los casos en que los nombres de los genes no estaban disponibles, se utilizaron en su lugar identificaciones de proteínas. Entre todas las muestras, algunas de las proteínas identificadas más abundantes fueron la apolipoproteína A y B (APOA, APOB), la albúmina (ALB), la alfa-2-macroglobulina (A2M), la cadena beta del fibrinógeno (FGB), la fibronectina (FN1), el complemento C3 (C3), la serotransferrina (LOC477072), el factor V de la coagulación (F5), la maltasa-glucoamilasa (MGAM) y varias proteínas no caracterizadas (LOC611458, LOC481722, A0A8I3P3U9). Para obtener una lista detallada de todas las proteínas identificadas y los datos brutos, consulte la Tabla Suplementaria S1.
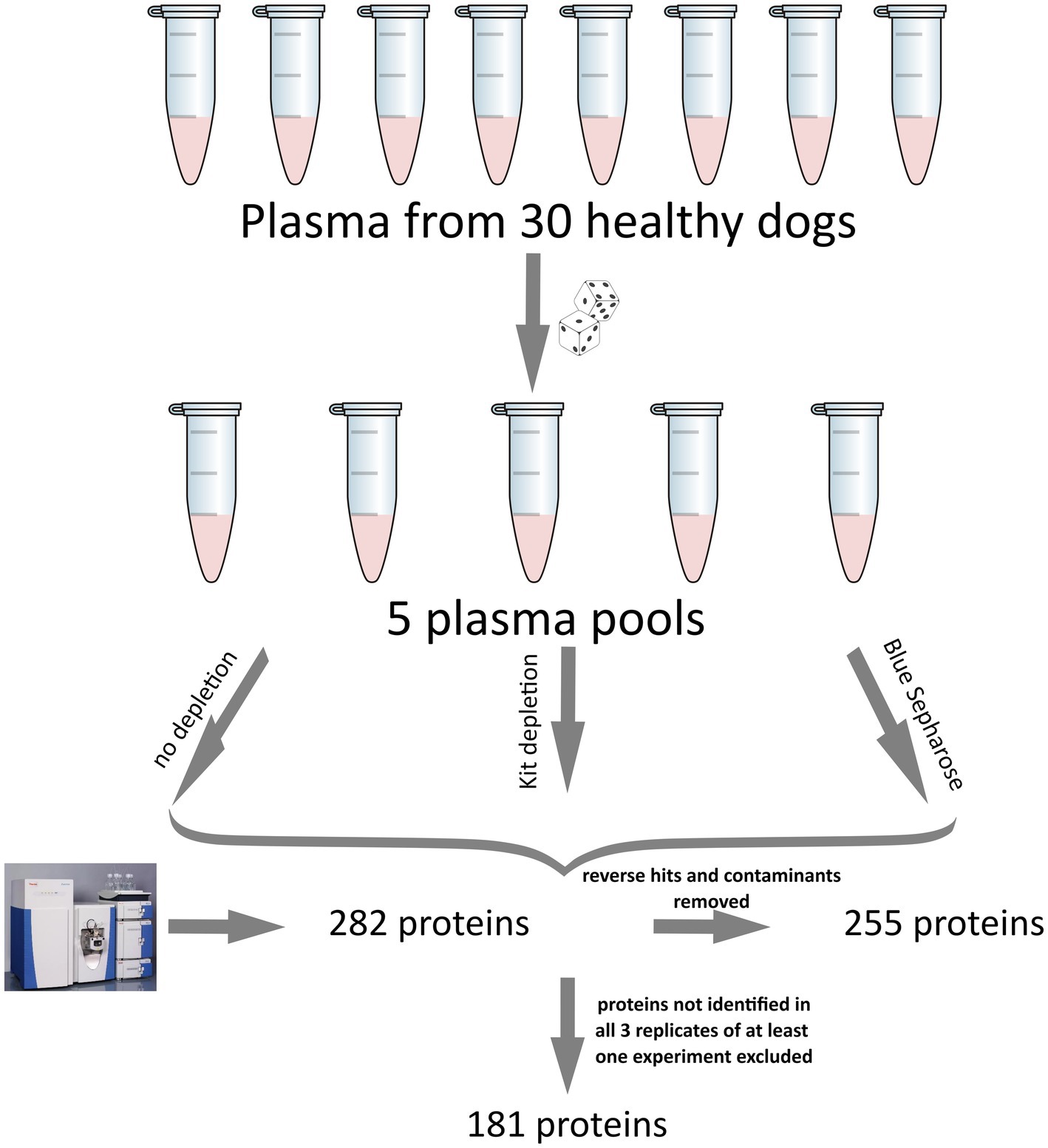
Antes de excluir las proteínas que no se detectaron en al menos tres réplicas de cualquiera de los tres grupos, 163 de las 181 proteínas estaban presentes en los tres grupos. Por el contrario, 15 proteínas solo estaban presentes en los grupos Control y Kit, cinco proteínas en los grupos Control y Blue-Sepharose, y 5 proteínas en los grupos Kit y Blue-Sepharose. Además, una proteína (S100A12) se identificó exclusivamente en el experimento de depleción del kit, y dos proteínas (proteínas que contienen dominios similares a Ig, ID de proteínas: A0A8I3P3T7 y A0A8I3P941) se identificaron únicamente en el experimento de depleción de Blue-Sepharose, y una proteína (AMBP) se identificó exclusivamente en el experimento sin depleción (control).
La clasificación de la abundancia de proteínas se puede observar en la Figura 2. El análisis de componentes principales demostró una segregación significativa y clara entre los tres métodos (Figura suplementaria S2). El agrupamiento jerárquico por pares y el análisis de correlación de todas las muestras de proteínas utilizando los dos métodos de depleción diferentes y el total de proteínas detectadas sin depleción como control, se representan en la Figura Suplementaria S3.
No se logró una depleción significativa de proteínas de alta abundancia, principalmente albúmina, con ninguna de las técnicas (Kit, Blue-Sepharose) en comparación con la técnica de control sin depleción (Figura suplementaria S1). Sin embargo, el cambio de pliegue log2 de la concentración de albúmina en las muestras después de usar el kit y la depleción de azul-sefarosa fue pequeño (0,312) pero estadísticamente significativo (q-valor = 0,007), siendo la albúmina más abundante después de la depleción con azul-sepharose. Una posible explicación para el agotamiento inadecuado de proteínas plasmáticas caninas de alta abundancia con el kit comercial es que el kit utilizado en este estudio está diseñado para agotar proteínas de alta abundancia en plasma o suero humano y no ha sido validado para agotar proteínas de plasma canino. Los anticuerpos utilizados para el kit no fueron completamente específicos y tienen afinidad por varias otras proteínas (27). Además, el método de depleción de Blue-Sepharose utilizado en este experimento aún no está estandarizado para muestras de animales, y planteamos la hipótesis de que las variaciones en el protocolo, como la cantidad de muestra o Blue-Sepharose añadido, podrían contribuir a mejores resultados de depleción. Un viejo estudio informó que la albúmina canina se une con una menor afinidad a la sefarosa azul. No pudimos notar esto antes de realizar este estudio porque el antiguo nombre de Blue Sepharose era Cibacron Blue (58).
En este estudio, utilizamos plasma canino en lugar de suero, ya que nuestro objetivo era identificar las proteínas involucradas en la cascada de coagulación. El fibrinógeno A (FGA) mostró una disminución significativa en la abundancia (log2 fold-change −1,469, q-valor = 0,006) después del agotamiento con el kit en comparación con el grupo control. Sin embargo, todavía no está claro hasta qué punto el fibrinógeno afecta a la depleción y cómo interfiere en la detección de proteínas menos abundantes.
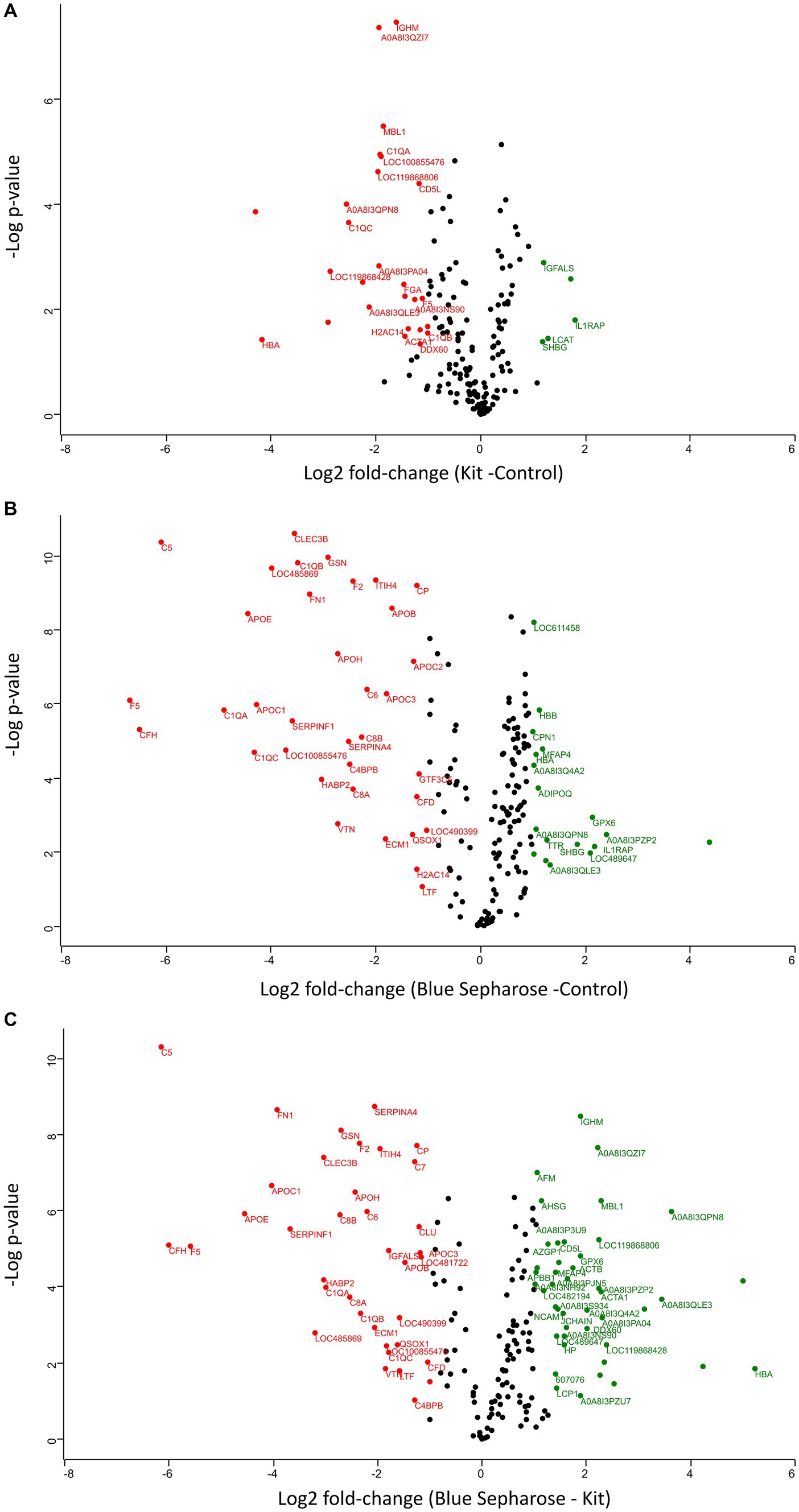
Los dos métodos de depleción diferentes muestran diferencias significativas en el cambio de pliegue de numerosas proteínas en comparación con las tres técnicas (Figuras 3A-C). Treinta y dos proteínas fueron diferencialmente abundantes entre los métodos de control y depleción del kit, siendo 27 más abundantes con el método de control y 5 con el método de depleción del kit (Tabla Suplementaria S2). Entre las proteínas más importantes que mostraron un aumento significativo después del agotamiento del kit en comparación con el método de control se encuentran la proteína accesoria del receptor de interleucina-1 (IL1RAP), la familia de transportadores de solutos 12 miembro 4 (LCAT), la subunidad lábil de ácido de la proteína de unión al factor de crecimiento similar a la insulina (IGFALS), la globulina fijadora de hormonas sexuales (SHBG) y la subunidad G de ATPasa de protón tipo V (A0A8I3PF02). Por otro lado, la subunidad alfa de hemoglobina (HBA), la ferritina (LOC119868428), el complemento C1q C (CIQC), la cadena alfa de fibrinógeno (FGA) y la proteína contenedora de dominio similar a Ig (A0A8I3PB96) fueron significativamente más abundantes en el experimento de control sin depleción (Figura suplementaria S4A). Cincuenta y tres proteínas fueron diferencialmente abundantes entre los métodos de control y depleción de Blue-Sepharose, siendo 35 más abundantes con el método de control y 18 con el método de depleción de Blue-Sepharose (Tabla suplementaria S3). Curiosamente, IL1RAP y SHBG mostraron un aumento significativo de la intensidad relativa después de la depleción con el método de Blue-Sepharose en comparación con el grupo de control (Figura suplementaria S4B). Finalmente, comparamos directamente las proteínas detectadas con las dos técnicas de depleción diferentes (Figura suplementaria S4C). Ochenta y dos proteínas fueron diferencialmente abundantes entre los métodos de agotamiento de Blue-Sepharose y kit, siendo 45 más abundantes con el método de Blue-Sepharose y 37 con el método de depleción de kit (Tabla suplementaria S4). Las proteínas más notables que fueron significativamente más abundantes después del agotamiento del kit incluyen el complemento C5 (C5), el factor V de la coagulación (F5), la apolipoproteína E (APOE), la fibronectina (FN1) y el miembro F de la familia de serpinas (SERPINF1), mientras que el HBA, la proteína contenedora de dominio similar a Ig (A0A8I3QPN8), la ferritina (LOC119868428), la proteína contenedora de dominio de lectina tipo C (MBL1) y la inmunoglobulina constante pesada mu (IGHM) se encontraron en concentraciones significativamente más altas utilizando el método Blue-Sepharose.
En nuestras condiciones experimentales, no encontramos un beneficio claro del uso de métodos de depleción porque uno de los objetivos del uso de estos métodos era aumentar el número de proteínas detectadas, lo que no se logró en nuestro estudio. Existe la posibilidad de que el kit para el agotamiento de proteínas no esté bien optimizado para el plasma de perros. Este parece ser el caso también de la azul-sepharose, ya que la albúmina no disminuyó significativamente y otras proteínas inesperadas disminuyeron, probablemente debido a una unión inespecífica. Por esta razón, con la actual configuración instrumental descrita aquí, consideramos que no es necesario, en principio, utilizar un método de depleción para el análisis de plasma canino, o alternativamente, se debe desarrollar y probar un nuevo método de depleción específico. En el caso de la azul-sefarosina, la disminución de la abundancia de proteínas específicas, pero no de albúmina, puede indicar una unión inespecífica e impredecible de estos conjuntos de proteínas. Por el contrario, la albúmina inesperadamente no disminuyó en concentración.
Un informe reciente indica la cuantificación de alrededor de 400 proteínas plasmáticas utilizando diferentes técnicas de fraccionamiento (59). También hemos utilizado recientemente con éxito la metodología descrita en este trabajo para descubrir la firma del proteoma plasmático del síndrome de diarrea hemorrágica aguda canina (AHDS). En ese estudio, detectamos un número de proteínas similar al reportado aquí. Por ejemplo, encontramos que la serpina3, la proteína de unión a lipopolisacáridos, la gliceraldehído-3-fosfato deshidrogenasa y el amiloide A sérico eran más abundantes en el plasma de perros afectados por AHDS. Por el contrario, otras proteínas, como la paraoxonasa, la selenoproteína, las amina oxidasas y la apolipoproteína C-IV, fueron significativamente menos abundantes. Se sabía que muchas proteínas identificadas y cuantificadas estaban asociadas con la inflamación, lo que valida la capacidad de este método para detectar candidatos a biomarcadores relacionados con enfermedades en medicina veterinaria (60).
Conclusión
El análisis de LC-MS mediante cuantificación sin marcadores puede detectar y cuantificar de forma fiable múltiples proteínas en el plasma canino, lo que lo convierte en una herramienta valiosa para desentrañar la patogénesis de diversas enfermedades en medicina veterinaria. Como herramienta, proporciona información influyente para el diagnóstico preciso de la enfermedad y la estimación del pronóstico en análisis futuros. Nuestros resultados indican que la depleción proteica con los dos métodos aquí descritos no se logra adecuadamente. Aunque la abundancia de varias proteínas puede diferir significativamente, estos métodos, tal como se realizan aquí, no contribuyen a la determinación de proteínas de menor abundancia en el plasma canino. Se espera que los estudios prospectivos controlados en modelos de enfermedades animales arrojen luz sobre la utilidad del análisis proteómico de espectrometría de masas LC sin etiquetas y sin gel en medicina veterinaria.
Declaración de disponibilidad de datos
Los datos presentados en el estudio se encuentran depositados en el repositorio Phaidra, número de acceso 2720; Los identificadores de prueba https://phaidra.vetmeduni.ac.at/o:2720 y se proporcionan más adelante como material complementario.
Declaración ética
El estudio en animales fue aprobado por la Comisión de Ética de la Universidad de Medicina Veterinaria. El estudio se llevó a cabo de acuerdo con la legislación local y los requisitos institucionales.
Contribuciones de los autores
PD: Conceptualización, Análisis formal, Investigación, Metodología, Software, Redacción – borrador original, Redacción – revisión y edición. BK: Curación de datos, Análisis formal, Metodología, Recursos, Software, Validación, Redacción – borrador original, Redacción – revisión y edición. CF: Curación de datos, Investigación, Redacción – borrador original, Redacción – revisión y edición. AR-R: Conceptualización, Curación de datos, Análisis formal, Investigación, Metodología, Administración de proyectos, Recursos, Software, Supervisión, Validación, Visualización, Redacción – borrador original, Redacción – revisión y edición. IB: Conceptualización, Obtención de fondos, Investigación, Metodología, Recursos, Supervisión, Validación, Visualización, Redacción – borrador original, Redacción – revisión y edición.
Financiación
El/los autor/es declara(n) que no se recibió apoyo financiero para la investigación, autoría y/o publicación de este artículo.
Reconocimientos
Para la espectrometría de masas, los autores desean agradecer la ayuda de la Core Facility BioSupraMol, respaldada por la Deutsche Forschungsgemeinschaft (DFG).
Conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un posible conflicto de intereses.
El autor o autores declararon ser miembros del comité editorial de Frontiers, en el momento de la presentación. Esto no tuvo ningún impacto en el proceso de revisión por pares ni en la decisión final.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o afirmación que pueda hacer su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fvets.2024.1356318/full#supplementary-material
Referencias
1. Fidock, M, y Desilva, B. Bioanálisis de biomarcadores para el desarrollo de fármacos. Bioanálisis. (2012) 4:2425–6. doi: 10.4155/BIO.12.253
2. Anderson, NL, Ptolomeo, AS, y Rifai, N. El enigma del diagnóstico de proteínas: ¿futuro sombrío o brillante? Clin Chem. (2013) 59:194–7. doi: 10.1373/CLINCHEM.2012.184705
3. Ahrens, CH, Brunner, E, Qeli, E, Basler, K y Aebersold, R. Generación y navegación de mapas de proteomas mediante espectrometría de masas. Nat Rev Mol Cell Biol. (2010) 11:789–801. doi: 10.1038/NRM2973
4. Cox, J y Mann, M. Proteómica cuantitativa de alta resolución para la biología de sistemas basada en datos. Annu Rev Biochem. (2011) 80:273–99. doi: 10.1146/ANNUREV-BIOCHEM-061308-093216
5. Bensimon, A, Heck, AJR y Aebersold, R. Proteómica basada en espectrometría de masas y biología de redes. Annu Rev Biochem. (2012) 81:379–405. doi: 10.1146/ANNUREV-BIOCHEM-072909-100424
6. Huang, Z, Ma, L, Huang, C, Li, Q y Nice, EC. Perfil proteómico del plasma humano para el descubrimiento de biomarcadores de cáncer. Proteómica. (2017) 17:1600240. doi: 10.1002/PMIC.201600240
7. Murphy, S, Zweyer, M, Mundegar, RR, Swandulla, D y Ohlendieck, K. Biomarcadores proteómicos séricos para enfermedades neuromusculares. Experto en Proteómica. (2018) 15:277–91. doi: 10.1080/14789450.2018.1429923
8. Ndao, M . Descubrimiento de biomarcadores en suero/plasma mediante espectrometría de masas de tiempo de vuelo de ionización por desorción láser mejorada en superficie (SELDI-TOF). Métodos Mol Biol. (2012) 818:67–79. doi: 10.1007/978-1-61779-418-6_5
9. Aebersold, R, y Mann, M. Exploración espectrométrica de masas de la estructura y función del proteoma. Naturaleza. (2016) 537:347–55. doi: 10.1038/NATURE19949
10. Geyer, PE, Holdt, LM, Teupser, D y Mann, M. Revisión del descubrimiento de biomarcadores mediante proteómica plasmática. Mol Syst Biol. (2017) 13:942. doi: 10.15252/MSB.20156297
11. Wild, D y Davies, C. Fundamentos del inmunoensayo en: El manual de inmunoensayo: teoría y aplicaciones de la unión de ligandos, ELISA y técnicas relacionadas. Elsevier. (2013). 1–26.
12. Vogeser, M, y Parhofer, KG. Cromatografía líquida por espectrometría de masas en tándem (LC-MS/MS): técnica y aplicaciones en endocrinología. Exp Clin Endocrinol Diabetes. (2007) 115:559–70. doi: 10.1055/S-2007-981458/ID/40
13. Adaszek, Ł, Banach, T, Bartnicki, M, Winiarczyk, D, Łyp, P y Winiarczyk, S. Aplicación de la técnica de espectrometría de masas MALDI-TOF para la detección de la infección por Babesia canis canis en perros. Parasitol Res. (2014) 113:4293–5. doi: 10.1007/s00436-014-4124-1
14. Escribano, D, Tvarijonaviciute, A, Kocaturk, M, Cerón, JJ, Pardo-Marín, L, Torrecillas, A, et al. Apolipoproteína-A1 sérica como posible biomarcador para el seguimiento del tratamiento de la leishmaniosis canina. Comp Immunol Microbiol Infect Dis. (2016) 49:82–7. doi: 10.1016/J.CIMID.2016.10.002
15. Locatelli, C, Piras, C, Riscazzi, G, Alloggio, I, Spalla, I, Soggiu, A, et al. Perfiles proteómicos séricos en CKCS con valvulopatía mitral. BMC Vet Res. (2016) 13:43. doi: 10.1186/s12917-017-0951-5
16. Martínez-Subiela, S, Horvatic, A, Escribano, D, Pardo-Marín, L, Kocaturk, M, Mrljak, V, et al. Identificación de nuevos biomarcadores para el seguimiento del tratamiento en leishmaniosis canina mediante análisis proteómico cuantitativo de alta resolución. Veterinario Immunol Immunopathol. (2017) 191:60–7. doi: 10.1016/J.VETIMM.2017.08.004
17. Franco-Martínez, L, Horvatić, A, Gelemanović, A, Samardžija, M, Mrljak, V, Contreras-Aguilar, MD, et al. Cambios en el proteoma salival asociados con la piometra canina. Frente Vet Sci. (2020) 7:277. doi: 10.3389/FVETS.2020.00277
18. Lucena, S, Coelho, AV, Muñoz-Prieto, A, Anjo, SI, Manadas, B, Capela E Silva, F, et al. Cambios en el proteoma salival de los perros beagle después de la pérdida de peso. Domest Anim Endocrinol. (2020) 72:106474. doi: 10.1016/J.DOMANIEND.2020.106474
19. Phochantachinda, S, Chantong, B, Reamtong, O, y Chatchaisak, D. Cambio en el proteoma plasmático asociado con el síndrome de disfunción cognitiva canina (CCDS) en Tailandia. BMC Vet Res. (2021) 17:60. doi: 10.1186/s12917-021-02744-w
20. Brummel, KE, Butenas, S, y Mann, KG. Estudio integrado del fibrinógeno durante la coagulación sanguínea. J Biol Chem. (1999) 274:22862–70. doi: 10.1074/JBC.274.32.22862
21. Profumo, A, Turci, M, Damonte, G, Ferri, F, Magatti, D, Cardinali, B, et al. Cinética de liberación de fibrinopéptido por trombina en función de la concentración de CaCl2: diferente susceptibilidad de FPA y FPB y evidencia de un efecto específico de la isoforma de fibrinógeno a la concentración fisiológica de Ca2+. Bioquímica. (2003) 42:12335–48. doi: 10.1021/BI034411E
22. Banks, RE, Stanley, AJ, Cairns, DA, Barrett, JH, Clarke, P, Thompson, D, et al. Influencias del procesamiento de muestras de sangre en el proteoma de bajo peso molecular identificado por espectrometría de masas de desorción/ionización láser mejorada en superficie. Clin Chem. (2005) 51:1637–49. doi: 10.1373/CLINCHEM.2005.051417
23. Anderson, NL, Polanski, M, Pieper, R, Gatlin, T, Tirumalai, RS, Conrads, TP, et al. El proteoma del plasma humano: una lista no redundante desarrollada por la combinación de cuatro fuentes separadas. Proteómica de células molares. (2004) 3:311–26. doi: 10.1074/MCP. M300127-MCP200
24. Omenn, GS, States, DJ, Adamski, M, Blackwell, TW, Menon, R, Hermjakob, H, et al. Resumen del proyecto del proteoma plasmático HUPO: resultados de la fase piloto con 35 laboratorios colaboradores y múltiples grupos analíticos, generando un conjunto de datos central de 3020 proteínas y una base de datos disponible públicamente. Proteómica. (2005) 5:3226–45. doi: 10.1002/PMIC.200500358
25. Deutsch, EW, Omenn, GS, Sun, Z, Maes, M, Pernemalm, M, Palaniappan, KK, et al. Avances y utilidad del proteoma del plasma humano. J Proteoma Res. (2021) 20:5241–63. doi: 10.1021/acs.jproteome.1c00657
26. Tu, C, Rudnick, PA, Martínez, MY, Cheek, KL, Stein, SE, Slebos, RJC, et al. Agotamiento de proteínas plasmáticas abundantes y limitaciones de la proteómica plasmática. J Proteoma Res. (2010) 9:4982–91. doi: 10.1021/PR100646W
27. Bellei, E, Bergamini, S, Monari, E, Fantoni, LI, Cuoghi, A, Ozben, T, et al. Depleción de proteínas de alta abundancia para el análisis proteómico sérico: eliminación concomitante de proteínas no dirigidas. Aminoácidos. (2011) 40:145–56. doi: 10.1007/S00726-010-0628-X
28. Cao, Z, Tang, HY, Wang, H, Liu, Q y Speicher, DW. Comparación sistemática de métodos de fraccionamiento para el análisis en profundidad de proteomas plasmáticos. J Proteoma Res. (2012) 11:3090–100. doi: 10.1021/PR201068B
29. Liu, T, Qiant, WJ, Gritsenko, MA, Xiao, W, Moldawer, LL, Kaushal, A, et al. Caracterización de alto rango dinámico del proteoma plasmático del paciente traumatizado. Proteómica de células molares. (2006) 5:1899–913. doi: 10.1074/MCP. M600068-MCP200
30. Pan, S, Chen, R, Crispin, DA, May, D, Stevens, T, McIntosh, MW, et al. Alteraciones proteicas asociadas con el cáncer de páncreas y la pancreatitis crónica encontradas en el plasma humano mediante perfiles proteómicos cuantitativos globales. J Proteoma Res. (2011) 10:2359. doi: 10.1021/PR101148R
31. Keshishian, H, Burgess, MW, Gillette, MA, Mertins, P, Clauser, KR, Mani, DR, et al. El flujo de trabajo cuantitativo multiplexado para el descubrimiento de biomarcadores sensibles en plasma produce nuevos candidatos para la lesión miocárdica temprana. Proteómica de células molares. (2015) 14:2375–93. doi: 10.1074/MCP. M114.046813
32. Herosimczyk, A, Dejeans, N, Sayd, T, Ozgo, M, Skrzypczak, WF y Mazur, A. Análisis del proteoma del plasma: geles y chips 2D. J Physiol Pharmacol. (2006) 57:81–93.
33. Desrosiers, RR, Beaulieu, É, Buchanan, M y Béliveau, R. Análisis proteómico de proteínas plasmáticas humanas mediante electroforesis en gel bidimensional y mediante matrices de anticuerpos tras el agotamiento de proteínas de alta abundancia. Bioquímica Celular Biophys. (2007) 49:182–95. doi: 10.1007/s12013-007-0048-z
34. Marcus, K, Lelong, C, y Rabilloud, T. ¿Qué espacio para la proteómica bidimensional basada en gel en un mundo de proteómica de escopeta? Protemas. (2020) 8:17. doi: 10.3390/PROTEOMES8030017
35. Agallou, M, Athanasiou, E, Samiotaki, M, Panayotou, G, and Karagouni, E. Identification of immunoreactive leishmania infantum protein antigens to asymptomatic dog sera through combined immunoproteomics and bioinformatics analysis. PLoS One. (2016) 11:e0149894. doi: 10.1371/JOURNAL.PONE.0149894
36. Franco-Martínez, L, Tvarijonaviciute, A, Horvatić, A, Guillemin, N, Bernal, LJ, Barić Rafaj, R, et al. Changes in saliva of dogs with canine leishmaniosis: a proteomic approach. Vet Parasitol. (2019) 272:44–52. doi: 10.1016/J.VETPAR.2019.06.014
37. Galán, A, Horvatić, A, Kuleš, J, Bilić, P, Gotić, J y Mrljak, V. El análisis LC-MS/MS del fosfoproteoma sérico de perro revela sitios de fosforilación nuevos y conservados: patrones de fosfoproteínas en la babesiosis causada por Babesia canis, un estudio de caso. PLoS Uno. (2018) 13:e0207245. doi: 10.1371/REVISTA. PONE.0207245
38. Winiarczyk, D, Michalak, K, Adaszek, L, Winiarczyk, M y Winiarczyk, S. Proteoma urinario de perros con lesión renal durante la babesiosis. BMC Vet Res. (2019) 15:439. doi: 10.1186/s12917-019-2194-0
39. Hormaeche, M, Carretón, E, González-Miguel, J, Gussoni, S, Montoya-Alonso, JA, Simón, F, et al. Análisis proteómico de la orina de perros infectados por Dirofilaria immitis. Parasitol veterinario. (2014) 203:241–6. doi: 10.1016/J.VETPAR.2014.01.025
40. Escribano, D, Cihan, H, Martínez-Subiela, S, Levent, P, Kocaturk, M, Aytug, N, et al. Cambios en las proteínas séricas en perros con infección por Ehrlichia canis. Microb Pathog. (2017) 113:34–9. doi: 10.1016/J.MICPATH.2017.10.024
41. Nabity, MB, Lees, GE, Dangott, LJ, Cianciolo, R, Suchodolski, JS, y Steiner, JM. Análisis proteómico de la orina de perros machos durante las primeras etapas de la lesión tubulointersticial en un modelo canino de enfermedad glomerular progresiva. Veterinario Clin Pathol. (2011) 40:222–36. doi: 10.1111/J.1939-165X.2011.00307.X
42. Chacar, F, Kogika, M, Sanches, TR, Caragelasco, D, Martorelli, C, Rodrigues, C, et al. Proteína urinaria de Tamm-Horsfall, albúmina, proteína de unión a la vitamina D y proteína de unión al retinol como biomarcadores tempranos de la enfermedad renal crónica en perros. Physiol Rep. (2017) 5:E13262. doi: 10.14814/PHY2.13262
43. Ferlizza, E, Isani, G, Dondi, F, Andreani, G, Vasylyeva, K, Bellei, E, et al. Proteoma y metaboloma urinario en perros (Canis lupus familiaris): el efecto de la enfermedad renal crónica. J Proteoma. (2020) 222:103795. doi: 10.1016/J.JPROT.2020.103795
44. Tvarijonaviciute, A, Gutiérrez, AM, Miller, I, Razzazi-Fazeli, E, Tecles, F, y Ceron, JJ. Un análisis proteómico del suero de perros antes y después de un programa de pérdida de peso controlado. Domest Anim Endocrinol. (2012) 43:271–7. doi: 10.1016/J.DOMANIEND.2012.04.004
45. Lucena, S, Varela Coelho, A, Anjo, SI, Manadas, B, Mrljak, V, Capela E Silva, F, et al. Análisis proteómico comparativo de la saliva de perros con y sin disfunción metabólica relacionada con la obesidad. J Proteoma. (2019) 201:65–72. doi: 10.1016/J.JPROT.2019.04.010
46. González-Arostegui, LG, Rubio, CP, Cerón, JJ, Tvarijonaviciute, A, y Muñoz-Prieto, A. Proteómica en perros: una revisión sistemática. Res Vet Sci. (2022) 143:107–14. doi: 10.1016/J.RVSC.2021.12.026
47. Yuan, C, Guo, Y, Ravi, R, Przyklenk, K, Shilkofski, N, Diez, R, et al. La proteína C de unión a miosina se fosforila diferencialmente tras el aturdimiento miocárdico en corazones de caninos y ratas, evidencia de nuevos sitios de fosforilación. Proteómica. (2006) 6:4176–86. doi: 10.1002/PMIC.200500894
48. Kjelgaard-Hansen, M, Christensen, MB, Lee, MH, Jensen, AL y Jacobsen, S. Isoformas de amiloide A sérico en suero y líquido sinovial de perros enfermos espontáneamente con enfermedades articulares u otras afecciones. Veterinario Immunol Immunopathol. (2007) 117:296–301. doi: 10.1016/J.VETIMM.2007.03.008
49. Plumb, RS, Rainville, PD, Potts, WB, Johnson, KA, Gika, E y Wilson, ID. Aplicación de cromatografía líquida-espectrometría de masas de ultra rendimiento para perfilar bilis de ratas y perros. J Proteoma Res. (2009) 8:2495–500. doi: 10.1021/PR801078A
50. Nakamura, K, Miyasho, T, Nomura, S, Yokota, H y Nakade, T. Análisis del proteoma del líquido cefalorraquídeo en beagles sanos y encefalitis canina. J Vet Med Sci. (2012) 74:751–6. doi: 10.1292/JVMS.11-0474
51. Lawrence, YA, Dangott, LJ, Rodrigues-Hoffmann, A, Steiner, JM, Suchodolski, JS, y Lidbury, JA. Análisis proteómico de tejido hepático de perros con hepatitis crónica. PLoS Uno. (2018) 13:e0208394. doi: 10.1371/REVISTA. PONE.0208394
52. Cerquetella, M, Rossi, G, Spaterna, A, Tesei, B, Gavazza, A, Pengo, G, et al. Análisis proteómico fecal en perros sanos y en perros que sufren diarrea sensible a los alimentos. Mundo Ciencia J. (2019) 2019:2742401. DOI: 10.1155/2019/2742401
53. Kuleš, J, de Torre-Minguela, C, Barić Rafaj, R, Gotić, J, Nižić, P, Ceron, JJ, et al. Biomarcadores plasmáticos de SIRS y MODS asociados a la babesiosis canina. Res Vet Sci. (2016) 105:222–8. doi: 10.1016/J.RVSC.2016.02.011
54. Tvarijonaviciute, A, Ceron, JJ, de Torre, C, Ljubić, BB, Holden, SL, Queau, Y, et al. Perros obesos con y sin disfunción metabólica relacionada con la obesidad: un enfoque proteómico. BMC Vet Res. (2016) 12:211. doi: 10.1186/s12917-016-0839-9
55. Rappsilber, J, Mann, M e Ishihama, Y. Protocolo para la micropurificación, enriquecimiento, prefraccionamiento y almacenamiento de péptidos para proteómica mediante StageTips. Nat Protoc. (2007) 2:1896–906. doi: 10.1038/nprot.2007.261
56. Tyanova, S, Temu, T, y Cox, J. La plataforma computacional MaxQuant para la proteómica de escopeta basada en espectrometría de masas. Nat Protoc. (2016) 11:2301–19. doi: 10.1038/nprot.2016.136
57. Tyanova, S, Temu, T, Sinitcyn, P, Carlson, A, Hein, MY, Geiger, T, et al. La plataforma computacional Perseus para el análisis exhaustivo de datos (prote)ómicos. Métodos Nat. (2016) 13:731–40. doi: 10.1038/nmeth.3901
58. Naval, J, Calvo, M, Lampreave, F, y Piñeiro, A. Interacciones de diferentes albúminas y sueros animales con azul de Cibacron insolubilizado. Evaluación de constantes de afinidad aparentes. Comp Biochem Physiol B. (1982) 71:403–7. doi: 10.1016/0305-0491(82)90401-1
59. Ravuri, HG, Noor, Z, Mills, PC, Satake, N y Sadowski, P. La adquisición independiente de datos permite una cuantificación robusta de 400 proteínas en plasma canino no agotado. Protemas. (2022) 10:9. doi: 10.3390/proteomas10010009
Palabras clave: proteómica plasmática, espectrometría de masas, canino, medicina veterinaria, biomarcador
Cita: Doulidis PG, Kuropka B, Frizzo Ramos C, Rodríguez-Rojas A y Burgener IA (2024) Caracterización del proteoma plasmático de perros adultos sanos. Frente. Vet. Sci. 11:1356318. doi: 10.3389/fvets.2024.1356318
Editado por:
Muhammad Saqib, Universidad de Agricultura, Faisalabad, Pakistán
Revisado por:
Nirajkumar Makadiya, Clínica Veterinaria Westbrook, Canadá
Abdulbaki Agbas, Universidad de Kansas City, Estados Unidos
Derechos de autor © 2024 Doulidis, Kuropka, Frizzo Ramos, Rodríguez-Rojas y Burgener. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia Creative Commons Attribution License (CC BY).
*Correspondencia: Alexandro Rodríguez-Rojas, Alexandro.Rojas@vetmeduni.ac.at; Iwan A. Burgener, Iwan.Burgener@vetmeduni.ac.at
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente a las de sus organizaciones afiliadas, o las del editor, de los editores y de los revisores. Cualquier producto que puede ser evaluada en este artículo o afirmación que puede ser hecha por su El fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía