
Comparación genómica entre Mycobacterium bovis y Mycobacterium microti

Comparación genómica entre Mycobacterium bovis y Mycobacterium microti y análisis in silico de biomarcadores basados en péptidos para el serodiagnóstico
 Charlotte Moens1,2*†
Charlotte Moens1,2*† Bert Bogaerts3†
Bert Bogaerts3† Víctor Lorente-Leal4
Víctor Lorente-Leal4 Kevin Vanneste3
Kevin Vanneste3 Sigrid C. J. De Keersmaecker3
Sigrid C. J. De Keersmaecker3 Nancy H. C. Roosens3
Nancy H. C. Roosens3 Laurent Mostin5 5
Laurent Mostin5 5 David Fretin1Sylvie Marché1
David Fretin1Sylvie Marché1- 1Laboratorio de Bacteriología Veterinaria, Departamento de Enfermedades Infecciosas Animales, Sciensano, Bruselas, Bélgica
- número arábigoLaboratorio de Bioquímica y Genética de Microorganismos, Instituto de Ciencia y Tecnología Biomolecular de Lovaina, Université Catholique de Louvain, Louvain-la-Neuve, Bélgica
- 3Actividades Transversales en Genómica Aplicada, Sciensano, Bruselas, Bélgica
- 4Centro de Vigilancia Sanitaria VISAVET, Universidad Complutense de Madrid, Madrid, España
- 5Centro Experimental Machelen, Sciensano, Machelen, Bélgica
En los últimos años, ha habido un aumento en el número de casos reportados de infección por Mycobacterium microti en varios animales, lo que puede interferir con el diagnóstico ante-mortem de la tuberculosis animal causada por Mycobacterium bovis. En este estudio, se utilizó la secuenciación del genoma completo (WGS) para buscar genes codificantes de proteínas para distinguir a M. microti de M. bovis. Además, se describe la estructura poblacional de los conjuntos de datos genómicos WGS de M. microti disponibles, incluidos tres nuevos aislados belgas de infecciones en alpacas. Los genes candidatos se identificaron mediante el examen de la presencia de las regiones de diferencia y mediante un análisis pangenómico de los datos disponibles de WGS. Un total de 80 genes mostraron variación de presencia-ausencia entre las dos especies, incluidos los genes que codifican las proteínas Prolina-Glutamato (PE), Prolina-Prolina-Glutamato (PPE) y Secuencia Polimórfica Rica en GC (PE-PGRS) involucradas en la virulencia y la interacción del huésped. El filtrado basado en la localización subcelular predicha, la homología de secuencia y la antigenicidad predicha dio como resultado 28 proteínas de 80 que se predijeron como antígenos potenciales. Dado que los péptidos sintéticos son menos costosos y variables que las proteínas recombinantes, se realizó un enfoque in silico para identificar epítopos lineales y discontinuos de células B en las proteínas seleccionadas. De las 28 proteínas, se identificaron 157 péptidos basados en epítopos de células B que discriminaban entre las especies de M. bovis y M. microti. Aunque todavía se requiere la confirmación mediante pruebas in vitro, estos péptidos sintéticos candidatos que contienen epítopos de células B podrían utilizarse en pruebas serológicas para diferenciar los casos de infección por M. bovis de M. microti, reduciendo así el diagnóstico erróneo en la vigilancia de la tuberculosis animal.
Introducción
La tuberculosis animal (TB) es una enfermedad infecciosa crónica causada por miembros del complejo Mycobacterium tuberculosis (MTBC), cuyos huéspedes naturales son mamíferos silvestres y domésticos (1). Mycobacterium bovis es la principal causa de tuberculosis en el ganado bovino (tuberculosis bovina, tuberculosis bovina) y es capaz de infectar a una amplia gama de otras especies animales, así como a los seres humanos (2). Por lo tanto, este patógeno zoonótico representa un alto riesgo para la salud animal y humana y para el comercio internacional de animales domésticos y productos de origen animal, lo que pone de manifiesto la necesidad de vigilancia.
El primer caso reportado de infección por M. bovis en una granja de alpacas en Bélgica ocurrió en 2015 (comunicación personal de M. Mori, Sciensano, Bélgica), lo que aumentó el interés en el diagnóstico ante-mortem en camélidos del Nuevo Mundo (NWC), como llamas y alpacas. Se ha informado de que las pruebas diagnósticas para la detección de la tuberculosis animal basadas en la inmunidad mediada por células (CMI), como la prueba de tuberculina intradérmica o el ensayo de IFNɤ, tienen poca precisión en las NWC (3, 4). Los ensayos de diagnóstico serológico pueden ser una alternativa prometedora, como lo demuestra (5). Durante la última década, se han desarrollado varios ensayos serológicos para detectar la tuberculosis animal en camélidos, como la prueba multiespecie ELISA INgezim Tuberculosis DR (Ingenasa, España) y la prueba multiplexada Enferplex Camelids TB (Enfer Scientific, Irlanda) (6, 7).
En 2020, se sospechó que las alpacas de dos granjas belgas diferentes, una de las cuales estuvo involucrada en el brote de tuberculosis animal de 2015, estaban infectadas con M. bovis sobre la base de resultados positivos en el diagnóstico serológico (INgezim Tuberculosis DR multiespecie y Enferplex Camelids TB). La necropsia de estos animales reveló lesiones típicas de la tuberculosis y la PCR en tiempo real (qPCR), realizada en los órganos y utilizando la secuencia de inserción IS6110 para la identificación de los miembros del MTBC, fue positiva. Sin embargo, la prueba de qPCR para diferenciar los miembros del MTBC, basada en polimorfismos del gen gyrB y realizada a partir de cultivo bacteriano (8), identificó M. microti (datos del Laboratorio Nacional de Referencia para la Tuberculosis Bovina, Sciensano, Bélgica).
Identificado por primera vez en ratones de campo, M. microti es otro miembro menos virulento de MTBC que puede infectar una amplia gama de especies animales y causar lesiones tuberculosas (9-11). Además, el impacto de la situación epidemiológica de M. microti en algunas áreas no parece ser irrelevante (11-13). Sin embargo, la Ley Europea de Sanidad Animal (UE 2016/429) no incluye a M. microti como agente etiológico de la tuberculosis animal, que según la legislación solo incluye a M. bovis, M. caprae y M. tuberculosis. En consecuencia, las infecciones por M. microti no son de declaración obligatoria y no dan lugar a las mismas medidas sanitarias (por ejemplo, retirada de animales, granjas bloqueadas) que las declaraciones de brotes de M. bovis o M. caprae. Por lo tanto, M. microti representa un riesgo de interferencia en el diagnóstico de la tuberculosis animal.
La investigación de biomarcadores para distinguir con precisión la infección por M. bovis de M. microti en el examen ante-mortem en animales ha recibido poca atención. Si bien las pruebas serológicas utilizadas para diagnosticar la tuberculosis animal en las NWC y otras especies, como el ganado, son efectivas para detectar animales infectados, no son lo suficientemente específicas como para distinguir entre la infección por M. bovis y M. microti (14-16). Por lo tanto, es importante identificar proteínas antigénicas específicas que puedan provocar una respuesta de anticuerpos y que puedan utilizarse en pruebas serológicas. Los epítopos de las células B son segmentos de proteínas antigénicas que pueden ser reconocidos por los anticuerpos (17), y se clasifican en epítopos lineales (es decir, secuencias continuas de aminoácidos) y conformacionales (es decir, estructuras tridimensionales plegadas). De particular interés es la identificación de epítopos específicos de células B que podrían usarse para construir péptidos sintéticos como biomarcadores.
Los miembros del MTBC tienen más del 99,9% de identidad de secuencia de ADN entre sí, pero difieren en fenotipo, virulencia y especie huésped. Las diferencias entre especies se limitan principalmente a deleciones de regiones de diferencia (RD), polimorfismos de un solo nucleótido (SNP) en genes codificadores de proteínas y regiones hipervariables. La tipificación molecular de los miembros de MTBC a menudo se realiza utilizando la poligotipia, un método basado en la reacción en cadena de la polimerasa que examina la deleción de secuencias espaciadoras en la región de repetición directa (DR) (18). Los perfiles de espoligotipos resultantes se pueden utilizar para comprender la diversidad genética y la relación de las cepas de MTBC. Se reportaron diversos espolagotipos para las cepas de M. microti (11), entre ellos, los más dominantes son los espoligotipos «tipo llama» y «tipo topillo», llamados así por los animales huéspedes en los que se observaron por primera vez (19). Los estudios han demostrado que las cepas de M. microti se caracterizan por regiones de deleción específicas de M. microti (MiD) junto con la deleción de parte de la región RD1 (RD1 mic), que incluye marcos de lectura abiertos (ORF) de genes de virulencia conocidos (20). Estas variaciones genómicas pueden ser útiles para la vigilancia epidemiológica y el diagnóstico de la tuberculosis animal (21-23). La organización genómica de los miembros del MTBC, principalmente M. bovis y M. tuberculosis, se ha explorado más a fondo utilizando la secuenciación del genoma completo (WGS), lo que permite una mejor comprensión de la patología y la identificación de biomarcadores para el diagnóstico y el tratamiento de la TB (24-27). Sin embargo, hasta la fecha, ha habido muy poca investigación sobre M. microti, y muy pocos conjuntos de datos de WGS están actualmente disponibles en bases de datos públicas.
Este estudio se inició con el objetivo de identificar posibles biomarcadores para distinguir M. microti de M. bovis mediante WGS. La información genómica se utilizó para detectar variaciones que afectaban a las proteínas y que podrían constituir la base de los métodos de diagnóstico serológico. Se utilizó un enfoque in silico para extraer epítopos de células B de las proteínas antigénicas putativas identificadas. Además, este estudio describe la diversidad genómica de M. microti basándose en datos disponibles públicamente, incluidas las cepas recientemente recolectadas de Bélgica.
Materiales y métodos
Cepas bacterianas y extracción de ADN
Se aislaron tres cepas de M. microti de los órganos de alpacas de granjas belgas. Dos de ellos fueron aislados en la misma granja a partir de animales con resultado positivo en las pruebas serológicas para tuberculosis animal (es decir, los aislados MI20-1 y MI20-2). Ambas alpacas presentaban lesiones tuberculosas en la necropsia. El tercer aislado se colectó de una alpaca que se encontró muerta (con lesiones tuberculosas) y originaria de otra granja (es decir, el aislado VAR696). No había un vínculo geográfico o epidemiológico aparente entre las dos granjas. Todas las muestras se recogieron en 2020.
Las cepas de M. microti MI20-1 y MI20-2 se cultivaron en un medio sólido de piedra o utilizando un sistema líquido automatizado BACTEC™ MGIT™ (Becton Dickinson, Sparks, MD, EE. UU.) en el Laboratorio Europeo de Referencia para la tuberculosis bovina en el Centro de Salud VISAVET (Madrid, España) y se incubaron a 37 °C durante varias semanas. M. microti VAR696 se procesó de manera similar en el Laboratorio Belga de Referencia para la tuberculosis bovina del Instituto Belga de Salud (Sciensano, Bruselas, Bélgica) y el ADN genómico se extrajo utilizando el kit QIAGEN DNeasy Blood and Tissue (Qiagen, Hilden, Alemania) de acuerdo con las instrucciones del fabricante para bacterias Gram-positivas con un paso de molienda mecánica previo para cumplir con los requisitos específicos para la extracción de ADN en el caso de micobacterias Spp. El ADN genómico de las cepas MI20-1 y MI20-2 se extrajo de un cultivo puro cultivado en medio Middlebrook 7H9 suplementado con albúmina oleica dextrosa catalasa (OADC) utilizando fenol-cloroformo-alcohol isoamílico (PCI) y luego se purificó utilizando etanol, como se describió previamente en (28). El pellet de ADN resultante se secó a temperatura ambiente y se volvió a suspender en 50-200 μL de agua destilada ultrapura (Sigma-Aldrich, St. Louis, MO, EE. UU.).
La calidad y la concentración del ADN se midieron mediante espectrofotometría utilizando el Nanodrop® 2000 (Thermo Fisher Scientific, Waltham, MA, USA) y la pureza del ADN se evaluó utilizando las proporciones A260/A280 y A260/A230.
La detección de los miembros del MTBC se realizó mediante una qPCR basada en el elemento IS6110 (29). La caracterización molecular de las micobacterias MTBC se realizó mediante qPCR basada en el polimorfismo del gen gyrB (8) y/o poligotipado de repetición variable directa (DVR) (30).
Secuenciación del genoma completo
Los extractos de ADN de los aislados MI20-1 y MI20-2 se enviaron al Laboratorio Nacional de Servicios Veterinarios en Ames, Iowa (EE. UU.) para su preparación en la biblioteca y secuenciación del genoma completo, mientras que el aislado VAR696 se secuenció en Sciensano (Bélgica). La preparación de la biblioteca se logró utilizando el kit de preparación de bibliotecas Nextera XT (Illumina, San Diego, CA, EE. UU.) de acuerdo con las instrucciones del fabricante. La secuenciación se llevó a cabo en ambos casos en un sistema MiSeq con la química V3, obteniendo lecturas de 250 pb en los extremos pareados, con el objetivo de una cobertura teórica de 60X basada en el tamaño del genoma esperado de ~ 4.4 Mbp de M. microti.
Preprocesamiento y montaje de novo
El preprocesamiento y el ensamblaje de novo se realizaron como se describió anteriormente (31). Brevemente, las lecturas sin procesar se recortaron usando Trimmomatic 0.38 (32) con las siguientes opciones: «LEADING» establecido en 10, «TRAILING» establecido en 10, «SLIDINGWINDOW» establecido en «4:20», «MINLEN» establecido en 40 e «ILLUMINACLIP» establecido en «NexteraPE-PE.fa:2:30:10». A continuación, las lecturas procesadas se ensamblaron de novo utilizando SPAdes 3.13.0 (33) con la opción «—cuidado» habilitada y «–cov-cutoff» establecido en 10. Los contigs menores de 1.000 pb se eliminaron utilizando la función «seq» de Seqtk 1.3 (disponible en https://github.com/lh3/seqtk). La calidad de los ensamblajes se evaluó utilizando QUAST 4.4 (34), con los ensambles filtrados como entrada. También se evaluaron varios controles de calidad adicionales incluidos en el flujo de trabajo: (1) detección de contaminantes utilizando Kraken 2 v2.0.7 (35); (2) evaluación de la mediana de la profundidad de secuenciación y la tasa de mapeo utilizando Bowtie 2 v2.4.1 (36) y SAMtools v1.9 (37); y (3) varios controles de calidad en las lecturas de entrada con FastQC v0.11.7 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). La Figura 1 ilustra el flujo del análisis bioinformático completo.
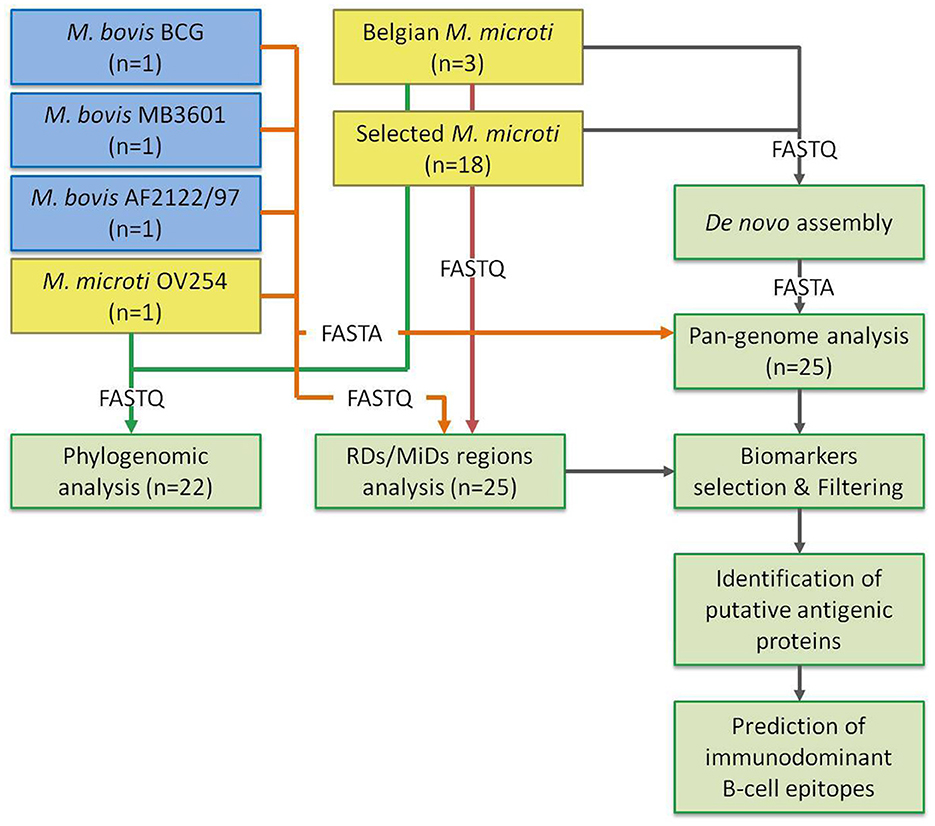
Análisis filogenómico
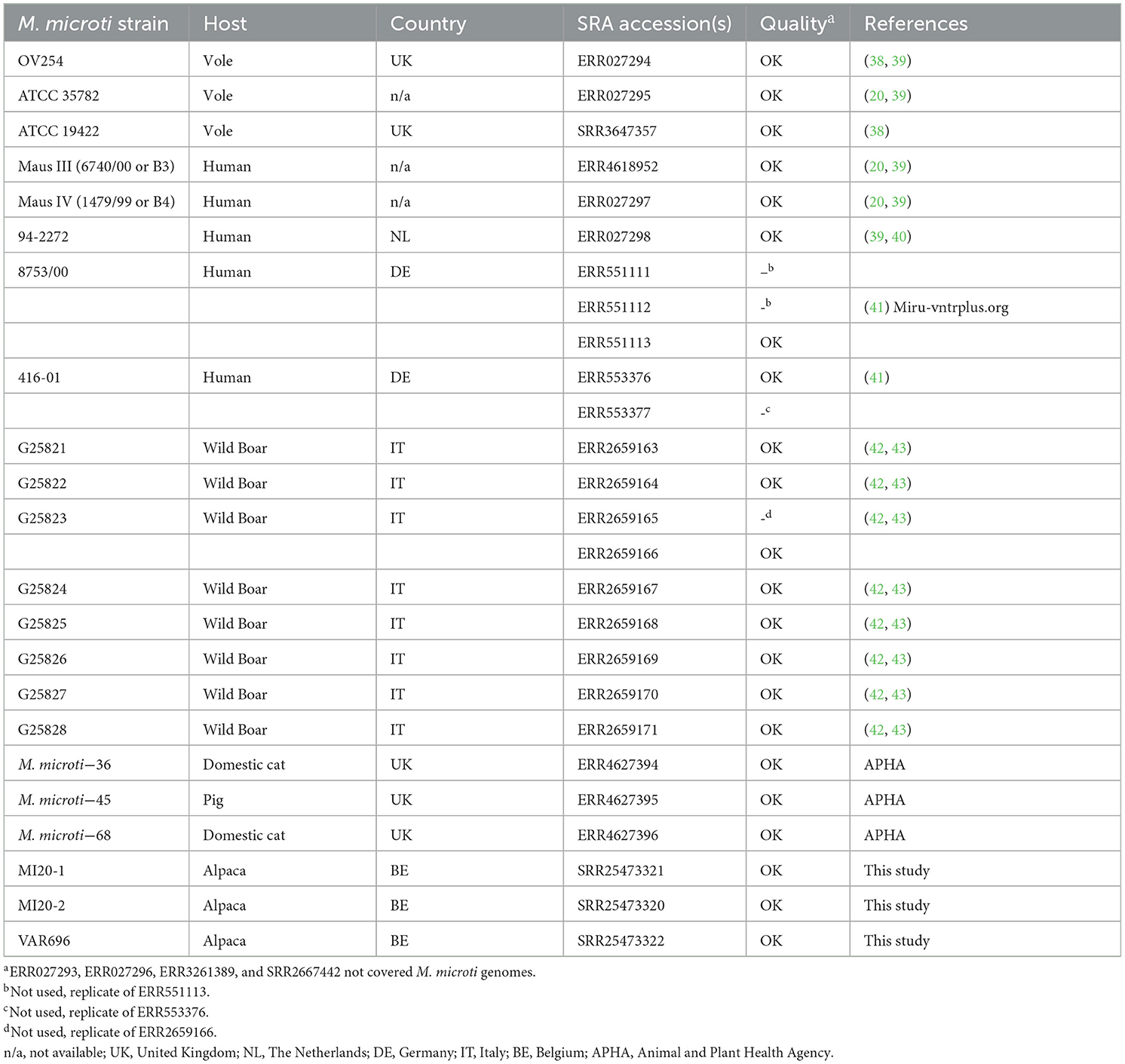
Se recuperaron todos los conjuntos de datos de M. microti disponibles en el Archivo de Lectura de Secuencias (SRA) del Centro Nacional de Información Biotecnológica (NCBI) que fueron generados por la secuenciación de Illumina (consultados el 24 de enero de 2022), para los cuales se proporciona una descripción general en la Tabla 1. Se disponía de tres conjuntos de datos para el aislado 8753/00, de los cuales se seleccionó ERR551113 por tener la mayor cobertura. El conjunto de datos ERR553377 también se excluyó del análisis, ya que se agrupó de manera idéntica en el análisis SNP con ERR553376, que se recopiló en el mismo estudio. Del mismo modo, el conjunto de datos ERR2659165 se excluyó del análisis, ya que es una réplica del conjunto de datos ERR2659166. Los conjuntos de datos con una cobertura estimada >100x, calculada sobre la base del número de bases en los archivos FASTQ y el tamaño del genoma del genoma de M. microti OV254 (acceso RefSeq GCF_904810325.1), se redujeron a ~100x utilizando la función «muestra» de seqtk 1.3. A continuación, la filogenia de los SNP se realizó utilizando PACU 0.0.5 (44), que utiliza BCFtools 1.17 (45) para la llamada de variantes y Gubbins 3.3.1 (46) para eliminar los SNP en regiones recombinantes. En primer lugar, las lecturas procesadas se asignaron al genoma OV254 de M. microti (acceso RefSeq GCF_904810325.1), utilizando la función PACU_map con el parámetro «–read-type» establecido en «illumine» y otras opciones dejadas en sus valores predeterminados. Los archivos BAM resultantes se utilizaron como entrada para el flujo de trabajo SNP de PACU. Se utilizó la herramienta en línea PHASTER (47) para generar un archivo BED con regiones de fagos (pro-) en el genoma de referencia y se proporcionó a la PACU con la opción «–ref-bed». La opción –use-mega’ se habilitó para realizar la selección automática de modelos y la construcción de árboles con MEGA X (48). Se seleccionó el modelo de mejor ajuste como modelo de sustitución con el menor valor de criterio de información bayesiano (BIC), seleccionando el modelo de 3 parámetros de Tamura. Los espoligotipos se determinaron in silico utilizando SpoTyping 2.1 (49) y se añadieron como anotaciones a la filogenia. Los valores de los parámetros «—min» y «—rmin» se establecieron en 1 para los conjuntos de datos con una cobertura estimada de < 30x, estimada en función de la asignación al genoma de referencia. Para otros conjuntos de datos, se utilizaron los valores de parámetro predeterminados.
Caracterización genómica de las regiones RDs y MiDs
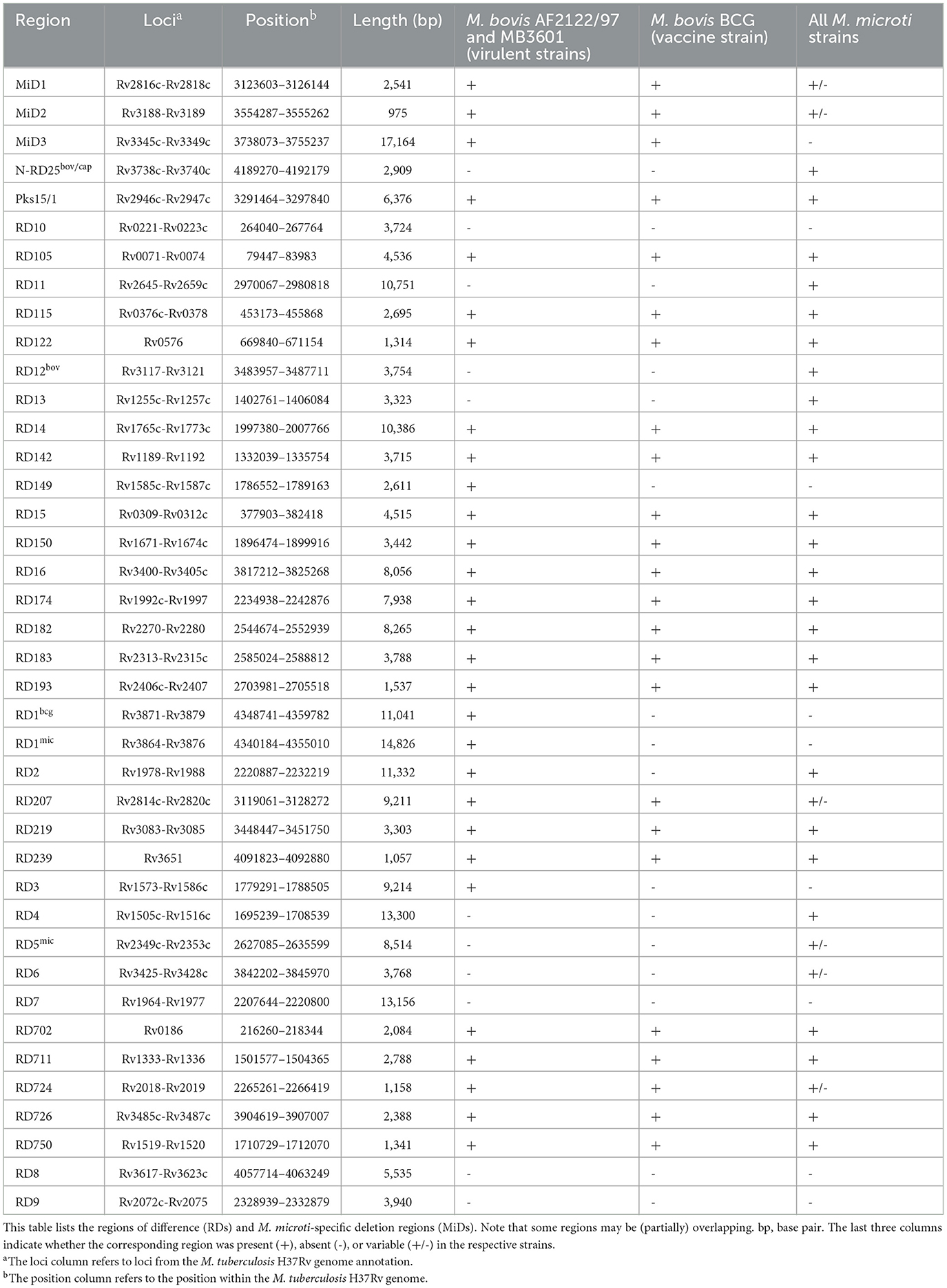
La presencia de RD y MiD se evaluó utilizando un enfoque basado en el mapeo de lecturas. La nomenclatura de RD y MiD utilizada en este estudio se basa en la de (20, 21, 50-52) y difiere de la propuesta por (53). En la Tabla 2 se ofrece una visión general de los RD y MiD evaluados. Se utilizó Bowtie2 2.4.1 con el preajuste «–very-sensitive-local» habilitado para mapear las lecturas procesadas a las secuencias de las regiones RD, obtenidas del genoma de referencia de M. tuberculosis H37Rv (acceso RefSeq NC_000962.3). El análisis incluyó los conjuntos de datos de Illumina FASTQ para los genomas de referencia M. bovis BCG Danish 1311 (SRR7983756), M. bovis AF2122/97 (ERR1744454), M. bovis MB3601 (ERR3825346) y M. microti OV254 (ERR027295) y todas las cepas de M. microti incluidas en el análisis filogenómico. Se consideró que las regiones que estaban cubiertas por al menos el 75% y con una profundidad media >5x estaban presentes.
Análisis del pangenoma
Para el análisis pangenómico se utilizó Roary. Se utilizaron tres cepas de referencia de M. bovis (es decir, la cepa de la vacuna BCG de M. bovis y las dos cepas virulentas de M. bovis AF2122/97 y MB3601) y una cepa de referencia de M. microti (OV254), para las cuales los genomas circulares se descargaron de NCBI RefSeq. Además, también se utilizaron los genomas ensamblados de las cepas de M. microti utilizadas en el análisis filogenómico, incluidos los de las tres cepas belgas. Se utilizó Prokka 1.14.6 (54) con opciones predeterminadas para la predicción de genes y la anotación funcional. Las proteínas de los genomas de referencia M. tuberculosis H37Rv, M. microti OV254, M. bovis AF2122/97 y M. bovis BCG Danish 1311 se proporcionaron con las opciones «-proteínas» como base para la anotación de genes. A continuación, se utilizó Roary 3.12.0 (55) para el análisis del pangenoma con la opción «—mafft» habilitada, la identidad mínima establecida en el 90% y los parálogos no divididos («-s»). Los gráficos UpSet se generaron utilizando el paquete de python UpSetPlot v0.8.0 (disponible en https://github.com/jnothman/UpSetPlot) para mostrar la superposición de genes entre especies y cepas. Los resultados del análisis del genoma pan se utilizaron para identificar genes codificantes de proteínas que podrían usarse como biomarcadores potenciales para la diferenciación de M. microti de M. bovis virulento (es decir, excluyendo la cepa de la vacuna danesa 1311 BCG de M. bovis).
Selección de posibles biomarcadores
Las proteínas, procedentes de los análisis RD/MiDs y pangenoma, presentes en las cepas virulentas de M. bovis (es decir, no en la cepa de la vacuna BCG de M. bovis) y ausentes en todas las cepas de M. microti, y viceversa, se seleccionaron como posibles biomarcadores. A continuación, se evaluaron de acuerdo con los siguientes tres criterios: localización subcelular, homología de secuencia y antigenicidad, como se detalla a continuación.
Predicción de la localización subcelular
SignalP 6.0 se utilizó para predecir la presencia de péptidos señal dependientes de sec y tat en proteínas (56). Estas secuencias cortas de aminoácidos N-terminales son reconocidas por la maquinaria clásica de translocación sec o tat y, por lo tanto, controlan la secreción y translocación de proteínas en las bacterias. SecretomeP 2.0 se utilizó para la predicción de proteínas secretadas no clásicas (57). Ambos programas están disponibles gratuitamente en el sitio web del Centro de Análisis de Secuencias Biológicas de la Universidad Técnica de Dinamarca (http://www.cbs.dtu.dk/services). Además, Psortb v3.0.3 [(58); https://www.psort.org/psortb/], Gpos-mPLoc [(59); http://www.csbio.sjtu.edu.cn/bioinf/Gpos-multi/] y CELLO2.5 [(60); http://cello.life.nctu.edu.tw/] se utilizaron para las predicciones de la localización subcelular de las proteínas candidatas. CELLO2.5 es un sistema de clasificación de máquinas de vectores de soporte (SVM) multiclase que utiliza cuatro tipos de esquemas de codificación de secuencia. Psortb3.0.3 utiliza una combinación de módulos, cada uno de los cuales se sabe que influye en la ubicación subcelular de las proteínas (por ejemplo, el número de hélices transmembrana, péptidos señalizadores, motivos de la membrana externa, …). Gpos-mPLoc identifica la localización subcelular de proteínas bacterianas Gram positivas mediante la fusión de la información de la ontología génica, así como la información del dominio funcional y la información de evolución secuencial. Las secuencias de aminoácidos se recuperaron de la base de datos de proteínas del NCBI y se utilizaron como entrada para todas las herramientas. También se consultó la base de datos UniProt (http://www.uniprot.org/) para obtener la localización subcelular de las proteínas.
Análisis de homología de secuencia
Biomarcadores putativos con alta similitud de secuencia con proteínas de miembros del complejo Mycobacterium avium-intracellulare (MAC; M. avium subespecie paratuberculosis, «M. avium subespecie hominissuis«, M. avium subespecie avium y M. intracellulare). Estas micobacterias ambientales se aíslan comúnmente del ganado y comparten proteínas con miembros de MTBC que pueden reaccionar de forma cruzada en el diagnóstico (61-63). Las similitudes de secuencia de todas las proteínas se evaluaron utilizando la base de datos de secuencias de proteínas no redundantes de la herramienta NCBI BLASTp (https://blast.ncbi.nlm.nih.gov/). Se descartaron las proteínas putativas con un porcentaje de identidad ≥55% y una cobertura de consulta ≥40% a las de la base de datos del NCBI.
Predicción de la antigenicidad
La antigenicidad de todas las proteínas se estimó utilizando ANTIGENpro (http://scratch.proteomics.ics.uci.edu/) y VaxiJen v2.0 (https://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). Las predicciones de ANTIGENpro se basan en un enfoque de dos pasos, que combina múltiples representaciones de la secuencia primaria y algoritmos de aprendizaje automático. Un clasificador final de la máquina de vectores de soporte (SVM) resume las predicciones resultantes y proporciona una probabilidad de si es probable que la proteína sea antigénica (64). VaxiJen 2.0 es un enfoque sin alineación para la predicción de antígenos, que se basa en la transformación automática de covarianzas cruzadas de secuencias de proteínas en vectores uniformes de propiedades de aminoácidos clave (65). Se utilizó un umbral de predicción de 0,5 para ambos programas, tal y como se recomienda en la literatura (64, 65). Las proteínas predichas como antigénicas por al menos una de estas dos herramientas se consideraron probablemente antigénicas.
Predicción de epítopos lineales y conformacionales de células B
La predicción de epítopos lineales y conformacionales de células B se realizó para todas las proteínas predichas como extracelulares, poco conservadas en micobacterias ambientales y antigénicas.
Para la predicción de epítopos lineales de células B, las secuencias de proteínas en formato FASTA se sometieron a cinco herramientas diferentes disponibles en la Base de Datos y Análisis de Epítopos (IEDB; https://www.iedb.org/), en base a las siguientes escalas: giro beta (66), accesibilidad superficial (67), flexibilidad (68), hidrofilicidad (69) y antigenicidad (70). Para cada herramienta, el tamaño de la ventana se estableció en siete aminoácidos y se utilizó el umbral predeterminado. BepiPred-2.0, disponible en el servidor IEDB, también se utilizó para predecir epítopos lineales de células B a partir de secuencias de aminoácidos en formato FASTA. La herramienta Bepipred 2.0 se basa en un algoritmo de bosque aleatorio entrenado con aminoácidos epítopos y no epítopos determinados a partir de estructuras cristalinas. Se utilizó el valor umbral predeterminado de 0,50 (71). Todos los resultados (es decir, todos los epítopos lineales de células B predichos) obtenidos con las herramientas IEDB se compararon para encontrar epítopos similares utilizando la herramienta Epitope Cluster Analysis v2.0 (http://tools.iedb.org/cluster/), con un umbral mínimo de identidad de secuencia del 70%. Los epítopos de consenso obtenidos con la herramienta Clúster de epítopos se seleccionaron como epítopos lineales finales de células B. La predicción de epítopos lineales de células B a través de múltiples herramientas y la agrupación de péptidos fue limitada para secuencias de proteínas con un alto número de aminoácidos (>1.500), PE_PGRS50 media (Rv3345c) y PPE55 (Rv3347c). En consecuencia, solo se utilizaron epítopos lineales antigénicos de células B predichos por el método de Kolaskar y Tongaonkar para estas proteínas.
Dado que la mayoría de los epítopos de las células B en las proteínas nativas son conformacionales (72), los epítopos conformacionales de las células B se predijeron utilizando el Discotope 3.0 (https://services.healthtech.dtu.dk/services/DiscoTope-3.0/) y ElliPro http://tools.iedb.org/ellipro/; (73) servidores. Las estructuras de las proteínas se descargaron de la base de datos PDB (https://www.rcsb.org/) o de la base de datos de estructuras de proteínas AlphaFold [https://alphafold.ebi.ac.uk/; (74)]. Se utilizaron los parámetros por defecto. Se conservaron las secuencias de aminoácidos, que oscilaban entre 8 y 25 residuos, que eran comunes a los epítopos lineales y conformacionales de las células B predichos. La antigenicidad de los epítopos de células B predichas como lineales y conformacionales se analizó con Vaxijen2.0. Los epítopos con un valor >0,5 se consideraron como los mejores péptidos epítopos de células B.
Disponibilidad de datos
Los conjuntos de datos que respaldan las conclusiones de este estudio se han depositado en el NCBI SRA bajo el número de acceso PRJNA1000766. En el Cuadro 1 se indican los números de accesión individuales.
Resultados
Preprocesamiento y calidad de los datos
En la Tabla Suplementaria 1 se proporcionan métricas de calidad de datos para el ensamblaje de novo y el análisis de mapeo de lectura de los conjuntos de datos generados internamente (es decir, los aislados belgas de M. microti). Los tres conjuntos de datos superaron los controles de calidad del flujo de trabajo (31), lo que indica que los datos eran adecuados para un análisis posterior.
Análisis filogenómico de los genomas disponibles de M. microti
De los 27 conjuntos de datos de M. microti recopilados del NCBI, 19 se incluyeron en el análisis filogenómico, y ocho se excluyeron debido a su baja calidad. Estos datos se complementaron con las tres cepas de M. microti secuenciadas en este estudio, resultando un total de 22 cepas de M. microti (Tabla 1). El resultado del análisis de máxima verosimilitud se muestra en la Figura 2 y las distancias de SNP por pares se muestran en la Tabla complementaria 2. Los aislados recogidos en este estudio muestran una similitud genómica muy alta, con solo ocho diferencias de SNP entre MI20-1 y MI20-2, y 18 SNPs entre estos dos aislados y VAR696. Sin embargo, parece poco probable que exista una relación epidemiológica directa entre estas cepas, dada la baja tasa de mutación de MTBC (75). La cepa más cercana recolectada de SRA fue M. microti-45, aislada de un cerdo en el Reino Unido, que difiere en 57 SNP de las tres cepas belgas en este estudio. La cepa M. microti-36, aislada de un gato doméstico en el Reino Unido, mostró 241-243 diferencias de SNP con las tres cepas belgas y 244 diferencias de SNP con M. microti-45. Si bien los valores de bootstrap para las ramas más largas fueron generalmente altos, los valores de bootstrap también indicaron una baja confianza en la posición relativa de estas ramas.
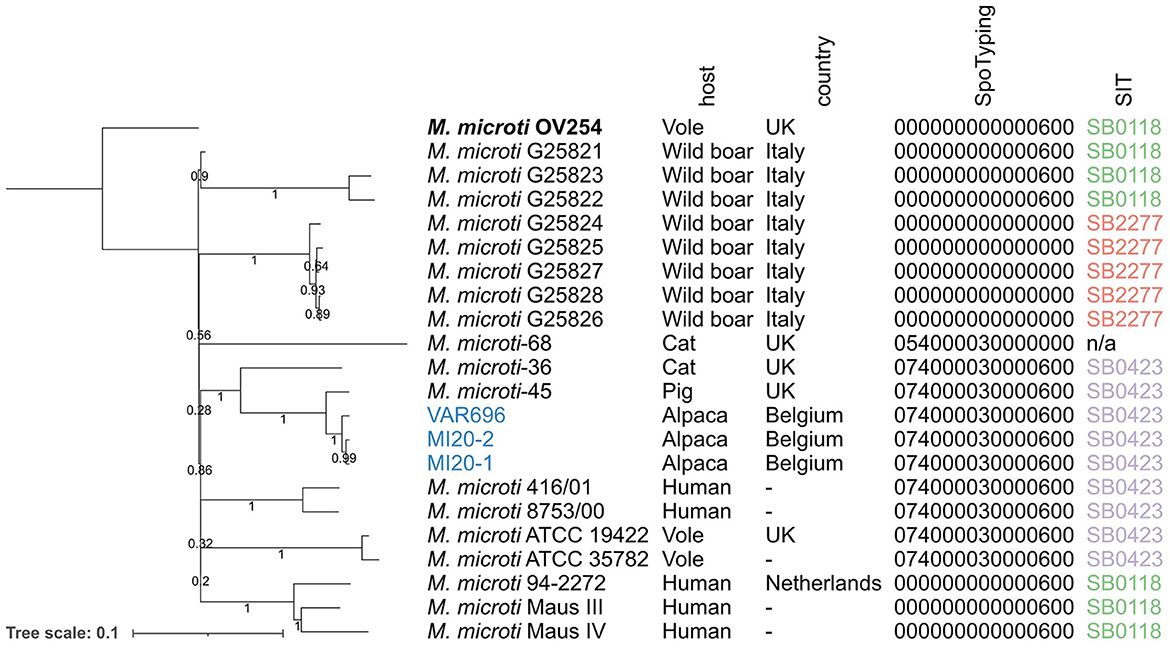
Figura 2. Filogenia de máxima verosimilitud de las cepas de M. microti colectadas en el NCBI y los aislados recolectados en este estudio. Las longitudes de las ramas y la barra de escala se expresan como sustituciones medias por sitio. El soporte de arranque se indica mediante los números en las ramas (proporciones basadas en 1.000 réplicas). M. microti OV254 se utilizó como genoma de referencia para la filogenia (resaltado en negrita). Los tres aislados recolectados como parte de este estudio se muestran en azul. Las anotaciones son (de izquierda a derecha): nombre de la cepa, especie huésped, país, espoligotipo determinado por SpoTyping en formato octal y número SIT. La filogenia estaba enraizada en el punto medio. Reino Unido, Reino Unido; N/A, no disponible; SIT, Spoligotype Tipo Internacional.
Las tres cepas belgas fueron asignadas al mismo espoligotipo que las cepas M. microti-45 y M. microti-36 en el mismo clado (el espoligotipo «tipo llama» SB0423). El mismo espoligotipo se observó en otros dos clados: (1) el clado portador de M. microti ATCC 19422 y ATCC35782, recolectado de ratones de campo, y (2) el clado portador de M. microti 416/01 y 8753/00, ambos aislados de humanos. La cepa M. microti-68 tenía un patrón espaciador similar, pero carecía de los espaciadores 37 y 38, lo que resultó en un espoligotipo diferente. Todos los demás aislados se asignaron a SB0118, conocido como el espoligotipo «tipo topillo», excepto el clado que contiene cinco aislados recolectados de jabalíes en Italia, que carecía de todos los espaciadores, asignándolos a SB2277.
Caracterización genómica de las regiones RDs y MiDs
Veinte de las 40 regiones RDs/MiDs analizadas estaban presentes en las tres cepas belgas y en las cepas de M. microti de referencia OV254, junto con las tres cepas de referencia de M. bovis estudiadas, como se muestra en la Figura 3. Las regiones RD7, RD8, RD9 y RD10 estuvieron (en su mayoría) ausentes en todas las cepas, excepto en M. tuberculosis H37Rv. Las regiones RD4, RD6, RD11 (profago phiRv2), RD12bov, RD13 y N-RD25bov/cap estuvieron ausentes en todas las cepas de M. bovis (cepas virulentas y vacunales), pero presentes en las cuatro cepas de M. microti. Sin embargo, ~60% (por debajo del umbral de detección del 75%) de la región N-RD25bov/cap estaba presente en las cepas de M. bovis, con el ORF PPE66 (Rv3738c) interrumpido y el locus PPE67 (Rv3739c) completamente ausente en estas cepas. No se encontraron ORFs intactos en las fracciones (< 25% de cobertura) de las regiones RD4, RD6, RD11 y RD13 que estaban presentes en las cepas de M. bovis. De manera similar, no se encontraron ORFs intactos para la fracción de la región RD12bov (~36% de cobertura) en las cepas de M. bovis, excepto para el locus Rv3117. Curiosamente, la regiónmicra RD5 estuvo en gran medida ausente de los genomas de M. bovis y M. microti OV254, pero estaba casi completamente cubierta por las tres cepas belgas de M. microti. A diferencia de M. microti OV254, los loci plcC (Rv2349c), plcB (Rv2350c), plcA (Rv2351c) y PPE38 (Rv2352c) estaban completamente cubiertos en las tres cepas belgas. La región MiD3 estuvo presente en todas las cepas de M. bovis estudiadas, pero estuvo por debajo del umbral de detección en todas las cepas de M. microti. Sin embargo, las cepas de M. microti contenían una gran fracción de la región MiD3, pero no ORFs intactos para PE_PGRS50 (Rv3345c), una proteína transmembrana (Rv3346c), PPE55 (Rv3347c) y una transposasa (Rv3349c). Solo la transposasa Rv3348c tenía un ORF intacto. Las regiones RD1mic, RD1bcg, RD3 y RD149 estuvieron presentes en M. bovis AF2122/97 y M. bovis MB3601, pero estuvieron ausentes en todas las demás cepas (cepas M. bovis BCG y M. microti). El comienzo de la regiónRD1 bcg (Rv3871-Rv3879c) estaba ausente en las cepas de M. microti, al igual que la mayor parte de la regiónmic RD1 (Rv3864-Rv3876). Para la región RD3 (profago phiRv1; Rv1573-Rv1586c), que se superpone parcialmente con la región RD149 (Rv1585c-Rv1587c), solo el ORF Rv1587c estaba intacto en las cepas de M. bovis BCG y M. microti. La región RD2 estuvo presente en todas las cepas, excepto en M. bovis BCG Danish 1311. Por último, las regiones RD207 y MiD1 parcialmente superpuestas estaban presentes en todas las cepas de MTBC excepto en M. microti OV254, lo que resultó en la ausencia o interrupción de CRISPR-Cas loci cas2 (Rv2816c), cas1 (Rv2817c), csm6 (Rv2818c) y csm5 (Rv2819c). Del mismo modo, la región MiD2 (Rv3188-Rv3189) estuvo presente en todas las cepas de MTBC analizadas, excepto en M. microti OV254.
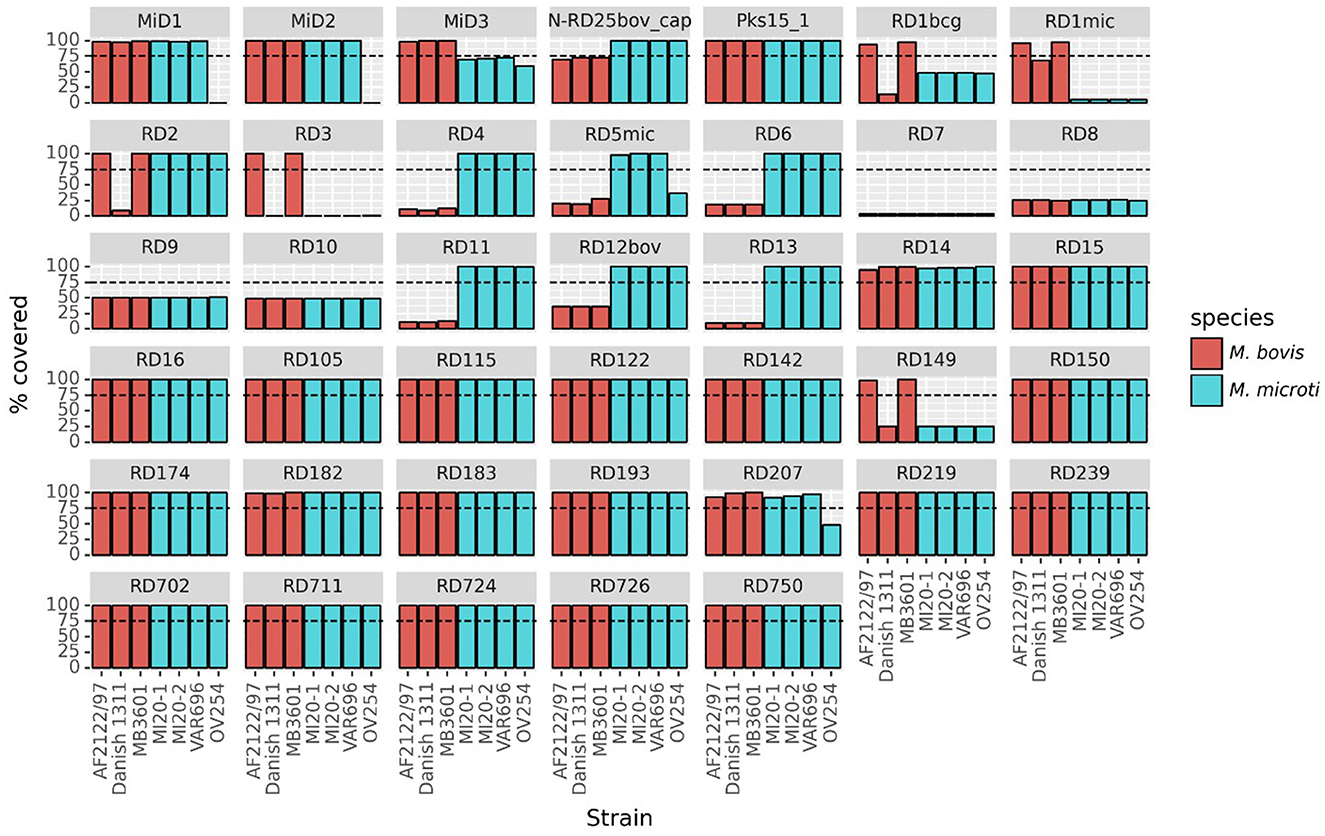
La presencia de los RD y MiD en todas las cepas de M. microti utilizadas en el análisis filogenómico (es decir, las 22 cepas de M. microti) se muestra en la Figura Suplementaria 1. Para la mayoría de las regiones analizadas, la cobertura fue muy similar entre todas las cepas de M. microti. Sin embargo, se observó cierta variabilidad entre las cepas para las regiones RD5mic y RD6, que estaban casi completamente cubiertas en las cepas belgas, pero parcialmente ausentes o ausentes en algunas otras cepas. La región MiD2 estuvo presente en todas las cepas de M. microti, excepto en la referencia OV254. Del mismo modo, la región RD724 estuvo presente en todas las cepas de M. microti excepto en la cepa G25822. Se observaron diferencias para los loci CRISPR-Cas (es decir, las regiones MiD1/RD207) que estaban completamente presentes en las cepas ATCC 35782, ATCC 19422, 416/01, 8753/00, microti-45 y tres cepas belgas, pero (parcialmente) ausentes en otras cepas, incluidas las cepas 94-2272, Maus III, Maus IV, microti-36, microti-68 y G25821-G25828 (es decir, los genes cas2 y cas1 ausentes y el csm6 gen interrumpido) y la cepa de referencia OV254. Si bien este análisis revela la presencia de regiones genómicas, no proporciona información sobre la expresión de los ORFs localizados en estas regiones, ya que pueden estar afectados por SNPs e indels.
Análisis pangenómico e identificación de señales de polimorfismo
El análisis del pangenoma detectó un total de 4.668 genes, de los cuales 1.401 fueron anotados como proteínas hipotéticas. En la Figura complementaria 2 se muestra una representación esquemática de la superposición entre especies. Para obtener genes que pudieran ser potenciales biomarcadores para diferenciar M. microti de M. bovis, se extrajeron los genes presentes exclusivamente en las dos cepas virulentas de M. bovis o en todas las cepas de M. microti (n = 22) estudiadas (Figura suplementaria 2). Obsérvese que, a efectos de este estudio, los resultados de las cepas danesas 1311 de M. bovis BCG no se incluyeron en este análisis, ya que esta cepa vacunal difiere de las cepas virulentas tradicionales. La selección resultante se muestra en la Tabla 3 y se realizó a partir de 47 y 34 genes exclusivamente presentes en cepas virulentas de M. bovis y M. microti, respectivamente. De estos 81 genes, 30 fueron anotados como proteínas hipotéticas (es decir, 19 genes exclusivos de M. bovis y 11 de M. microti).
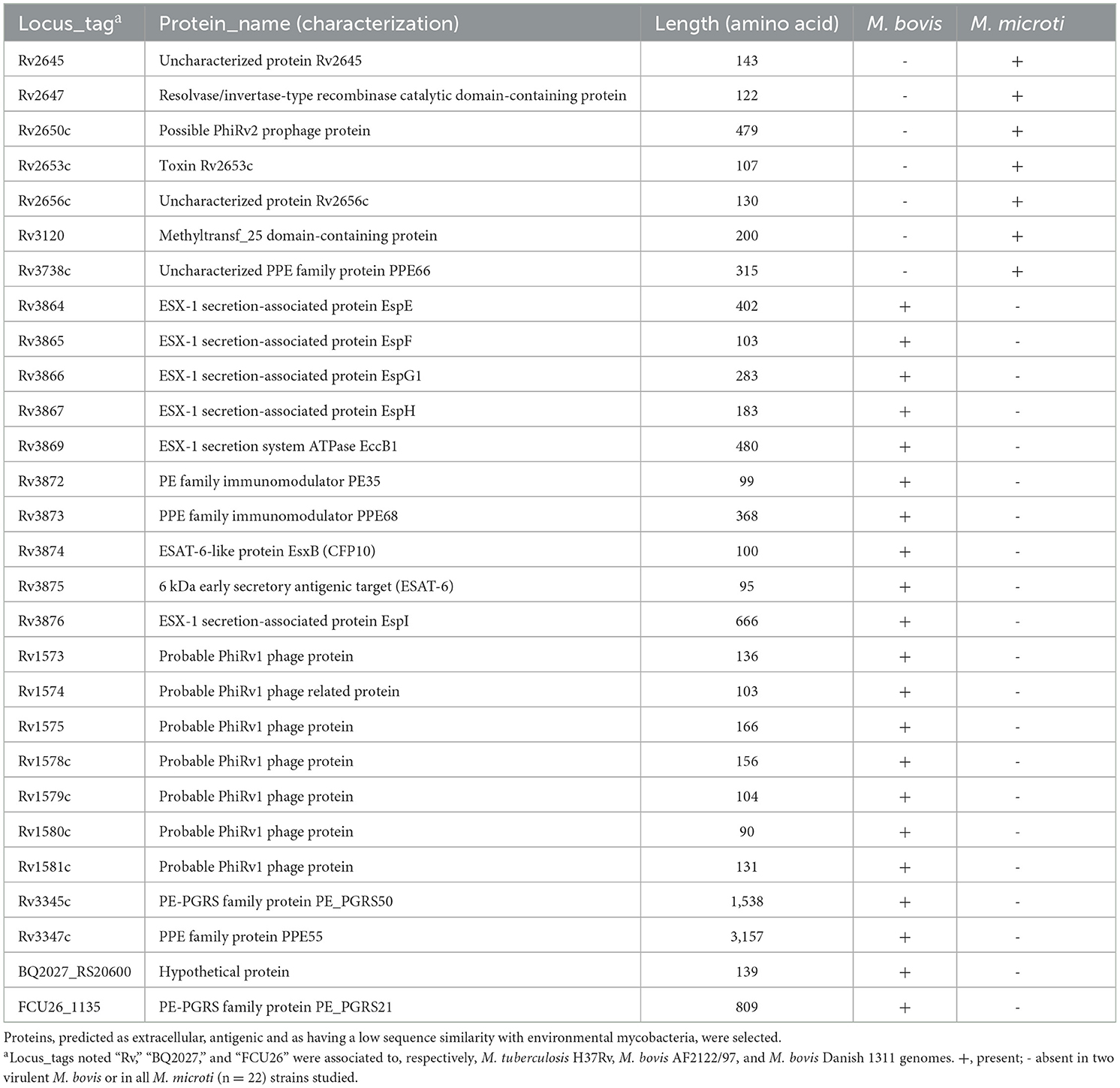
Entre los 47 genes exclusivamente presentes en el virulento M. bovis (incluidos los ORF localizados en las regionesRD1 mic, MiD3 y RD3) había varios genes que codifican para las proteínas prolina-glutamato (PE) y prolina-prolina-glutamato (PPE), así como las proteínas EsxR (Rv3019c) y EsxS (Rv3020c). Esta deleción, correspondiente a la región MiD4 (Rv3018c-Rv3022c), se reportó previamente en la cepa OV254 de M. microti y otras cepas de M. microti aisladas de pacientes (40). Otros genes codificadores de proteínas, como el PE_PGRS21 (FCU26_1135), la hipotética proteína Rv0530A (locus_tag BQ2027_RS02780), una transposasa (BQ2027_RS17270) y la hipotética proteína (WP_015631092.1; locus_tag BQ2027_RS20600) también estuvieron ausentes en las 22 cepas de M. microti estudiadas y presentes en las cepas virulentas de M. bovis (Tabla 3, punto A).
Además de los genes asociados con las regiones RD4, RD11, RD12bov y RD13 (en su mayoría ausentes en M. bovis), se identificaron otros tres genes adicionales entre 34 genes que estaban exclusivamente presentes en todos los M. microti. Estos genes codifican la proteína hipotética Rv1508A, una proteína hipotética (JN986_RS12585) y la proteína de membrana integral Rv3162c. Investigaciones posteriores revelaron que el gen Rv3162c estaba presente en los genomas de M. bovis, pero contenía una deleción de un solo nucleótido que daba lugar a un producto génico inactivo (Tabla 3, punto B). Cabe destacar que algunos loci registrados como eliminados en cepas de M. microti o M. bovis en el pangenoma son probablemente artefactos relacionados con el método de Roary utilizado en este trabajo, que se basa en genomas y anotaciones ensamblados (imperfectos). Otros genes estaban presentes en múltiples copias, con diferentes variantes, y su presencia o ausencia no pudo ser confirmada.
Además, investigamos las diferencias entre las cepas de M. microti estudiadas en este trabajo, centrándonos en las tres cepas belgas y la cepa de referencia OV254. Se encontró que tres ORFs (ORF2-ORF4) fueron eliminados o interrumpidos en las cepas belgas de M. microti. Estos se asociaron con la región RvD2 que incluye plcD, una glicosiltransferasa (ORF2), una sulfito oxidasa (ORF3) y un transportador de la familia RND (ORF4), que se sabe que está ausente en la cepa H37Rv de M. tuberculosis (región de deleción relacionada con H37Rv o RvD). La región adyacente a RvD2, llamada RD152, estaba completamente ausente en estas tres cepas. También se encontró que estas dos regiones estaban parcialmente (RvD2) o completamente (RD152) ausentes en la cepa M. microti-45, la cepa filogenéticamente más cercana de las tres cepas belgas. Además, se encontró que la región RD152 estaba ausente en la cepa G25824. Se encontró que otros genes estaban deledos en las tres cepas belgas, pero presentes en las cepas OV254 y M. bovis (Tabla 3, punto C). Además de los genes asociados a RDs/MiDs, exclusivamente ausentes en M. microti OV254, se identificaron otros cinco genes durante el análisis del pangenoma, como se muestra en la Tabla 3, punto D.
Identificación de genes codificantes de proteínas como potenciales marcadores de diagnóstico
Proteínas codificadas por los genes de RDs presentes en cepas virulentas de M. bovis y ausentes en todas las cepas de M. microti estudiadas (RD1mic, RD3 y MiD3) y viceversa (RD4, RD11, RD12bov, RD13 y N-RD25bov/cap), así como las proteínas con variación de presencia/ausencia entre M. bovis virulento y M. microti Se analizaron más a fondo las cepas. Dado que el objetivo era identificar antígenos «universales», no se consideraron los genes que presentaban variación de presencia/ausencia entre las diferentes cepas de M. microti.
De estas 80 proteínas, se predijo que 52 tenían un péptido señal, que sería secretado de forma no clásica y/o que se localizaría en la matriz extracelular mediante las herramientas SignalP6.0, SecretomeP2.0, Psortb3.0, Gpos-mPLoc y CELLO2.5. Protein-BLAST reveló que 27 de estas 52 proteínas tenían poca o ninguna homología con ninguna secuencia de miembros MAC en la base de datos. Tres proteínas pertenecientes a la proteína de la familia PE/PPE (PE_PGRS50, PPE55 y PE_PGRS21) mostraron una similitud de secuencia entre el 40 y el 60%, pero una cobertura de consultas muy baja (5-18%) en las secuencias alineadas, y se mantuvieron en este trabajo. A continuación, se evaluó la propensión antigénica de estas 30 proteínas utilizando los servidores VaxiJen 2.0 y ANTIGENpro.
Después de estos pasos de selección, quedaron un total de 28 proteínas que fueron predichas por al menos una de las herramientas para ser secretadas o localizadas en la superficie celular, con baja conservación en micobacterias ambientales y propiedades antigénicas putativas (Tabla 4 y Tablas Suplementarias 3-5).
Predicción de epítopos serodominantes
Se identificaron un total de 157 regiones predichas como epítopos lineales y conformacionales con una puntuación de antigenicidad >0,5 en el servidor VaxiJen 2.0 y se consideraron como epítopos prometedores para el serodiagnóstico. Las secuencias peptídicas con mayor puntuación de antigenicidad se muestran en la Tabla 5 para las 28 proteínas analizadas.

Tabla 5. Lista final de los epítopos de las células B del análisis in silico.
Discusión
M. microti es menos virulento en los seres humanos y el ganado que otros miembros del MTBC (51), y no se considera un agente causal de tuberculosis en animales según la nueva ley de sanidad animal (UE 2016/429). Sin embargo, su propagación y el creciente número de casos notificados de infección en diferentes huéspedes (9-11, 13, 76, 77) es motivo de preocupación para la identificación y el tratamiento de estrategias de control de la tuberculosis en animales, especialmente cuando la prevalencia de la tuberculosis animal es baja. Los datos de campo sugieren que M. microti puede interferir con varias pruebas diagnósticas ante-mortem utilizadas en los programas de vigilancia de la tuberculosis animal en ganado, camélidos y otras especies [datos no publicados del NRL belga; (11)].
En este estudio, primero realizamos la secuenciación del genoma completo en tres cepas de campo de M. microti y comparamos la diversidad genómica dentro de los datos de la secuenciación del genoma completo de M. microti disponibles públicamente. En segundo lugar, comparamos los genomas completos de diferentes cepas de M. microti y M. bovis, mediante la caracterización de polimorfismos de secuencia grande (RDs, MiDs y RvD) y análisis pangenómicos para identificar marcadores potenciales para discriminar entre las dos micobacterias en el diagnóstico de TB animal. El análisis se centró en las proteínas que probablemente sean adecuadas para las pruebas diagnósticas serológicas. Los ensayos serológicos son las principales pruebas ante-mortem utilizadas para detectar la TB en camélidos [debido al bajo rendimiento de los ensayos IFNɤ y SICCT en estos huéspedes; (4)] sin embargo, actualmente no hay ensayos disponibles para distinguir entre las infecciones por M. microti y M. bovis. A continuación, se utilizó un enfoque in silico para seleccionar los mejores epítopos de células B a partir de estas proteínas para su uso en el serodiagnóstico.
El análisis filogenómico proporcionó información sobre la diversidad genética dentro de las especies de M. microti, incluidas las diferencias entre los aislados recolectados de diferentes especies hospederas. Como se mostró anteriormente en (39), las cepas de M. microti tendían a agruparse en función de su patrón de espoligopatrón más que de su especie huésped. Sin embargo, el soporte de arranque para las relaciones entre clados fue muy bajo, lo que indica una baja confianza en la ubicación relativa de los clados, posiblemente debido a los limitados datos de WGS disponibles para M. microti. Se optó por armonizar la detección del espoligotipo dedeterminándola in silico utilizando los conjuntos de datos de WGS para reducir el sesgo entre las muestras. En algunos casos, los espoligotipos que se extrajeron de los datos de la WGS difirieron ligeramente de los informados anteriormente (20, 38), lo que puede deberse a la dificultad inherente para determinar los espoligotipos, tanto en técnicas de laboratorio seco como de laboratorio húmedo. Las distancias de SNP entre cepas de M. microti aisladas de granjas de alpaca belga podrían proporcionar una indicación de posibles vínculos epidemiológicos entre cepas. Se ha estimado que la tasa media de sustitución de M. tuberculosis es de 0,3 a 0,5 SNP por genoma por año y se han definido umbrales de SNP que van desde 5 (estricto, lo más probable) hasta 12 SNP (más putativo) para inferir eventos de transmisión (78, 79). Varios estudios llevados a cabo en aislados de M. bovis sugieren que una tasa similar podría ser aplicable para especies micobacterianas adaptadas a los animales (78, 80-83). Sobre la base de estos umbrales de SNP, es poco probable que las cepas de M. microti VAR696 y MI20-1, MI20-2, aisladas de dos granjas belgas diferentes y que difieren en 18 SNP, estén relacionadas epidemiológicamente. En contraste, las dos cepas de M. microti MI20-1 y MI20-2 aisladas de la misma granja y que muestran 8 SNPs de diferencia, podrían estar relacionadas con un evento de transmisión pasado o interacciones epidemiológicas pasadas dentro de la misma granja. Sin embargo, estos perfiles de SNP también podrían deberse a diferentes introducciones anteriores, a la introducción de animales infectados (con diferentes genotipos) procedentes de diferentes localidades, o a diferentes cepas que circulan en la misma explotación o en la región.
Las variaciones genómicas, como RD o SNP, son marcadores utilizados para estudiar la evolución o la relación epidemiológica existente entre las especies del MTBC o las cepas de la misma especie (82, 84). Estas variaciones también pueden ser útiles como marcadores diagnósticos. En este estudio, las diferencias genómicas entre las cepas de M. microti probadas se investigaron primero comparando los RD y los MiD. Se observó poca variación entre las cepas de M. microti para la mayoría de las regiones analizadas. Como se informó anteriormente, las cepas de M. microti se caracterizan por la deleción de la regiónmicra RD1, que contiene varios genes que codifican el sistema de secreción ESX-1, incluidos los factores de virulencia ESAT6 (Rv3875) y CFP10 (Rv3874). Las regiones MiD3, MiD4 y RD3 también estuvieron ausentes en todas las cepas de M. microti analizadas, incluidas las cepas belgas de M. microti (20, 39, 40). Sin embargo, se observaron diferencias entre las diferentes cepas de M. microti para algunos RDs/MiD, como RD5mic, RD6, RD207/MiD1 y MiD2. La región RD5 contiene los loci de fosfolipasa C (plcA, plcB y plcC), y es muy variable entre las diferentes especies de micobacterias tuberculosas, pero también entre cepas de la misma especie, como se muestra en (21) y (39). La región RD6, que contiene el elemento IS1532, también se describe como altamente variable entre las cepas de MTBC (21). La presencia o deleción (parcial) de la región RD207 (que incluye la región MiD1) en las cepas de M. microti da como resultado la pérdida de espaciadores en el locus DR y, por lo tanto, estaría asociada con los espoligotipos de llama y topillo observados (20, 85). Dado que el cribado de RD se limita a regiones del genoma de M. tuberculosis H37Rv, se realizó un análisis pangenómico para revelar diferencias adicionales. Este análisis reveló la ausencia de varios genes que, después de una investigación más detallada, se identificaron como asociados con las regiones RvD2 y RD152. Estas dos regiones solo se encontraron (parcialmente) delecionadas en las cepas belgas y M. microti-45, pero estaban completamente presentes en la OV254 de referencia y otras cepas de M. microti estudiadas (excepto en la región RD152 que también se encontró delecionada en la cepa G25824). La región RvD2 se encuentra aguas abajo del gen plcD (Rv1755c) y codifica tres ORF (Mb1785-Mb1787) que se predice que son una proteína hipotética, una sulfito oxidasa putativa y una proteína transportadora transmembrana putativa (86-88). Se sabe que esta región está ausente en M. tuberculosis H37Rv, pero está presente en otros miembros del MTBC como M. bovis. La región RD152 codifica los genes Rv1758-Rv1765c (89), incluyendo una cutinasa putativa (cutinasa) y un miembro de la familia de proteínas PE_PGRS (wag22). Las deleciones de la región RD152 se han identificado previamente en las cepas nueve del linaje de M. africanum (90) y en las cepas de M. tuberculosis Beijing/W (91). Hasta donde sabemos, estas deleciones nunca han sido reportadas en M. microti u otras especies de MTBC adaptadas a los animales. Ambas regiones han sido reportadas como puntos calientes para las secuencias de inserción de IS6110, lo que puede facilitar eventos de recombinación (20, 85, 92). Por lo tanto, un mecanismo de deleción mediado por IS6110 podría ser responsable de la pérdida de las regiones RvD2 y RD152 en estas cepas. Sin embargo, las cepas con estas deleciones aún eran capaces de causar lesiones de tuberculosis en alpacas, como se observó con las cepas belgas de M. microti, lo que sugiere que estas regiones pueden no ser esenciales para la virulencia.
Se seleccionaron genes que mostraban variación de presencia-ausencia como posibles marcadores proteicos para distinguir las infecciones por M. bovis de las infecciones por M. microti. Se identificaron un total de 80 genes codificantes de proteínas que estaban exclusivamente presentes en todas las cepas de M. microti analizadas y ausentes en todas las cepas virulentas de M. bovis analizadas, o viceversa. El método de detección génica empleado en el análisis del pangenoma se basó, en algunos casos, en ensamblajes de genomas fragmentados. Por lo tanto, es posible que los genes se hayan pasado por alto debido a fracturas de ensamblaje u otras imprecisiones en los genomas ensamblados o predicciones de genes, lo que pone de relieve la necesidad de verificación in vitro. Sin embargo, este tipo de análisis son un método de cribado potente y rápido que puede utilizarse para generar bibliotecas de péptidos que se pueden probar in vitro. Muchos biomarcadores potenciales identificados en el presente análisis pangenómico han sido reportados previamente como antígenos potenciales para mejorar el diagnóstico serológico de la tuberculosis humana y/o bovina: Rv3872 (PE35), Rv3874 (CFP10), Rv3875 (ESAT6), Rv3876 (EspI) en RD1 (93-96), Rv1573, Rv1577c en RD3 (97), o Rv1508c, Rv1509, Rv1514c, Rv1516c en RD4 (93, 97). Además, varios de los genes con variación de presencia-ausencia identificados en este estudio codifican proteínas pertenecientes a las familias PE, PPE y Secuencia Polimórfica Rica en GC (PE_PGRS), incluyendo PE_PGRS21, PPE55 y PE_PGRS50. Las proteínas de estas familias, que constituyen ~10% del genoma de M. bovis (22), han sido descritas como factores de virulencia implicados en la patogénesis de la tuberculosis, pero también como factores que tienen un papel potencial en la variabilidad antigénica dentro del MTBC (98, 99). Además, se ha reportado que varias proteínas de la familia PE/PPE/PE_PGRS son inmunogénicas y pueden ser útiles en el diagnóstico de tuberculosis humana y/o animal (98, 100). Cabe destacar que los antígenos ESAT6 y CFP10, que se utilizan en varios ensayos para el diagnóstico de la tuberculosis en animales (por ejemplo, el ensayo IFNɤ y la prueba de tuberculosis Enferplex Camelid), están ausentes en las cepas de M. microti, pero presentes en las cepas de M. bovis (7, 101, 102). En consecuencia, la ausencia de una respuesta de anticuerpos a estos antígenos no confirma la ausencia de infección por M. microti, lo que pone de manifiesto la importancia de los antígenos específicos de M. microti.
El principal problema en el establecimiento de sistemas de inmunodiagnóstico es la identificación de proteínas inmunogénicas específicas de las especies objetivo. Se realizaron varios análisis in silico adicionales para obtener un conjunto de proteínas inmungénicas potenciales óptimas, incluida una evaluación de la homología de secuencia, la probabilidad antigénica y la accesibilidad de los anticuerpos. Las proteínas con alta similitud/identidad de secuencia con micobacterias ambientales, como los miembros de la MAC comúnmente aislados del ganado, pueden reaccionar de manera cruzada en las pruebas serológicas y, por lo tanto, se excluyeron (61, 63). Después de este filtrado adicional, se retuvieron un total de 28 proteínas candidatas. Sin embargo, es posible que los antígenos efectivos hayan sido eliminados por este filtrado. Por ejemplo, no se predijo que proteínas como Rv3871 (103), Rv1509, Rv2658c (97) y Rv1586c (104) fueran secretadas/localizadas en la superficie celular y/o antigénicas en este trabajo, mientras que se ha informado que inducen una respuesta de anticuerpos en pacientes con tuberculosis. Dado que nuestro objetivo era maximizar la detección de casos de infección por M. bovis vs. M. microti, solo se consideraron los antígenos proteicos «universales» presentes en todas las cepas de M. microti estudiadas, pero ausentes en todas las cepas virulentas de M. bovis (y viceversa). Aunque los genes que estaban ausentes/presentes en las cepas belgas de M. microti (y algunas otras cepas) no se consideraron como antígenos potenciales en este trabajo, puede ser valiosa una mayor investigación de estas proteínas.
La producción de proteínas recombinantes para obtener antígenos micobacterianos es una tarea desafiante asociada con altos costos y variabilidad de antígenos relacionados con los sistemas de expresión y los pasos de purificación de proteínas recombinantes. El uso de péptidos sintéticos puede proporcionar uniformidad en las preparaciones de antígenos y, por lo tanto, en los ensayos (105). Por lo tanto, la identificación de péptidos que contengan epítopos de células B inmunodominantes puede ser esencial para el desarrollo de estrategias diagnósticas dirigidas a la detección de anticuerpos. Una ventaja adicional es la reducción del coste asociado al número de péptidos para los que se debe evaluar experimentalmente la reactividad de los anticuerpos. Muchos estudios se han centrado principalmente en epítopos lineales de células B, mientras que una fracción importante de los epítopos de células B son conformacionales (106). Hasta hace poco, la predicción de epítopos conformacionales de células B requería que la estructura de la proteína se depositara en la base de datos PDB o el uso de herramientas de modelado de homología (106, 107). Sin embargo, el recientemente lanzado AlphaFold basado en redes neuronales es una poderosa alternativa que permite una predicción altamente precisa de la estructura de las proteínas (74). En este trabajo, se buscaron péptidos basados en epítopos de células B lineales y conformacionales utilizando varias herramientas in silico disponibles en línea para predecir epítopos de células B en función de secuencias de aminoácidos y / o datos estructurales de proteínas (74, 108). Por lo tanto, se identificaron un total de 157 péptidos que contenían epítopos de células B predichos a partir de las 28 proteínas seleccionadas previamente utilizando el enfoque in silico. Curiosamente, se informó previamente que algunos péptidos, que se derivaron de la secuencia completa de las proteínas candidatas, como el péptido EISTNIRQAGVQYSRADEEQ para RV3874 (109) o el péptido AGMKLGWHPYHFPDEPDSKQ para Rv2653c (110), inducen una respuesta CMI de moderada a fuerte en animales tuberculosos o pacientes, lo que sugiere que estos péptidos podrían activar tanto la inmunidad humoral como la celular y tienen potencial en diversas aplicaciones de diagnóstico. Sin embargo, no se encontró información en la literatura científica sobre el reconocimiento de estos péptidos específicos por parte de los anticuerpos en un ensayo serológico.
Los ensayos in silico pueden guiar las pruebas experimentales para detectar antígenos putativos de manera más eficiente. En este trabajo, se utilizó un enfoque in silico para identificar péptidos antigénicos putativos que contienen epítopos de células B que discriminan entre infecciones por M. bovis y M. microti en el serodiagnóstico de tuberculosis animal. No obstante, se requieren análisis in vitro para identificar péptidos inmunológicamente relevantes entre los seleccionados in silico. La inmunorreactividad de los péptidos debe evaluarse mediante inmunoensayo (por ejemplo, ELISA) con un número suficiente de sueros de referencia, es decir, sueros de animales presuntamente no infectados con un estado conocido y sueros de animales con infección por M. bovis o M. microti confirmada por cultivo bacteriano y métodos moleculares. Los péptidos con los valores más altos de sensibilidad y especificidad se evaluarán más a fondo para determinar su capacidad serodiagnóstica en un panel más grande de sueros. Datos anteriores informaron que la respuesta de anticuerpos a los antígenos puede aumentar en bovinos, alpacas y cabras después de la prueba de tuberculina intradérmica, lo que puede afectar el rendimiento de las pruebas serológicas, en particular la sensibilidad (4, 14, 109). Esto debería ser considerado en los estudios futuros. Además, aunque el uso de péptidos sintéticos cuidadosamente seleccionados disminuye la inclusión de regiones de reacción cruzada de los antígenos proteicos, podría ser necesario confirmar la ausencia de una respuesta específica en el ensayo utilizando sueros de animales infectados por NTM. Dado que las combinaciones de antígenos micobacterianos (péptidos o proteínas recombinantes) están indicadas para mejorar el serodiagnóstico de la tuberculosis en humanos y animales (16, 105, 111), el rendimiento diagnóstico de los péptidos confirmados como inmunogénicos en ensayos preliminares podría evaluarse más a fondo en ensayos serológicos multiplexados o mediante antígenos polipeptídicos.
Con el uso de WGS, pudimos extraer biomarcadores potenciales en todo el genoma. Además, el análisis basado en SNP del genoma completo nos permitió caracterizar las relaciones entre cepas. A medida que aumenta el número de conjuntos de datos de la secuenciación del genoma completo de M. microti, a lo que contribuye nuestro estudio, la eficacia de estos análisis basados en la secuenciación del genoma completo puede aumentar aún más. En conclusión, los resultados presentados en este estudio pueden contribuir al desarrollo de pruebas serológicas que puedan diferenciar entre M. bovis y M. microti en el futuro, evitando así diagnósticos erróneos en el control de TB en animales.
Declaración de disponibilidad de datos
Los conjuntos de datos presentados en este estudio se pueden encontrar en repositorios en línea. Los nombres de los repositorios y los números de acceso se pueden encontrar en el artículo/Material complementario.
Declaración ética
No se requirió la aprobación ética para el estudio con animales de acuerdo con la legislación local y los requisitos institucionales porque los órganos de las alpacas se obtuvieron como parte del programa nacional belga de control de la tuberculosis bovina (TBB), de acuerdo con las directrices oficiales de la Autoridad Federal para el control de la TBB en Bélgica [es decir, la Agencia Federal para la Seguridad de la Cadena Alimentaria [FASFC] y los servicios veterinarios].
Contribuciones de los autores
CM: Conceptualización, Curación de datos, Análisis formal, Investigación, Metodología, Software, Redacción – borrador original, Redacción – revisión y edición. BB: Conceptualización, Curación de datos, Análisis formal, Investigación, Metodología, Software, Redacción – borrador original, Redacción – revisión y edición. VL-L: Escritura – revisión y edición. KV: Metodología, Supervisión, Redacción – revisión y edición. SD: Metodología, Redacción – revisión y edición. NR: Escritura – revisión y edición. LM: Escritura, revisión y edición. DF: Redacción – revisión y edición. SM: Conceptualización, Supervisión, Redacción – revisión y edición.
Financiación
El/los autor/es declara(n) que no se recibió apoyo financiero para la investigación, autoría y/o publicación de este artículo.
Reconocimientos
Agradecemos a Wiebke Jansen y Philippe Vannoorenberghe por su asistencia técnica, junto con la Agencia de Sanidad Animal y Vegetal (APHA) por la información sobre las cepas de M. microti aisladas de animales en el Reino Unido. También agradecemos a los técnicos del servicio de actividades transversales en Genómica Aplicada en Sciensano, Bélgica, por realizar una de las corridas de secuenciación del genoma completo. Además, nos gustaría agradecer al Laboratorio Europeo de Referencia para la Tuberculosis Bovina por su asistencia en la detección de la infección por parte del MTBC.
Conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un posible conflicto de intereses.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o afirmación que pueda hacer su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fvets.2024.1446930/full#supplementary-material
Figura complementaria 1 | Presencia de regiones RDs y MiDs en los conjuntos de datos de M. microti. En esta figura se muestra el porcentaje de las regiones RD y MiDs correspondientes que se cubrieron con el enfoque basado en mapeo de lectura para cada conjunto de datos.
Figura complementaria 2 | Resultados del análisis pangenómico, que muestran el número de genes detectados exclusivamente en todas las cepas de M. microti y todas las cepas virulentas de M. bovis. Nótese que se consideraron las 22 cepas de M. microti y las dos cepas virulentas de M. bovis (AF2122/97 y MB3601). Las barras horizontales indican el número total de genes detectados en las especies correspondientes mediante el análisis del pangenoma. Las líneas verticales indican el tamaño de la superposición, tal como lo indican los puntos en la matriz de pertenencia.
Referencias
1. WOAH. Infección por el complejo Mycobacterium tuberculosis. En: Organización Mundial de Sanidad Animal—Código Sanitario para los Animales Terrestres, Capítulo 8.12. París (2023).
2. Palmer MV, Thacker TC, Waters WR, Gortázar C, Corner LAL. Mycobacterium bovis: un patógeno modelo en la interfaz del ganado, la vida silvestre y los seres humanos. Vet Med Int. (2012) 2012:E236205. doi: 10.1155/2012/236205
Resumen de PubMed | Texto completo de Crossref | Google Académico
3. García-Bocanegra I, Barranco I, Rodríguez-Gómez IM, Pérez B, Gómez-Laguna J, Rodríguez S, et al. Tuberculosis en alpacas (Lama pacos) causada por Mycobacterium bovis. J Clin Microbiol. (2010) 48:1960–4. doi: 10.1128/JCM.02518-09
Resumen de PubMed | Texto completo de Crossref | Google Académico
4. Rhodes S, Holder T, Clifford D, Dexter I, Brewer J, Smith N, et al. Evaluación de pruebas de interferón gamma y anticuerpos antituberculosos en alpacas. Clin Vac Immunol. (2012) 19:1677–83. doi: 10.1128/CVI.00405-12
Resumen de PubMed | Texto completo de Crossref | Google Académico
5. Lyashchenko KP, Greenwald R, Esfandiari J, Meylan M, Burri IH, Zanolari P. Respuestas de anticuerpos en camélidos del Nuevo Mundo con tuberculosis causadas por Mycobacterium microti. Microbiol veterinario. (2007) 125:265–73. doi: 10.1016/j.vetmic.2007.05.026
Resumen de PubMed | Texto completo de Crossref | Google Académico
6. Bezos J, Casal C, Álvarez J, Díez-Guerrier A, Rodríguez-Bertos A, Romero B, et al. Evaluación de la realización de pruebas diagnósticas celulares y serológicas para el diagnóstico de tuberculosis en un hato de alpacas (Vicugna pacos) infectado naturalmente con Mycobacterium bovis. Ant: Vet Med. (2013) 111:304–13. doi: 10.1016/j.prevetmed.2013.05.013
Resumen de PubMed | Texto completo de Crossref | Google Académico
7. Krajewska-Wedzina M, Didkowska A, Sridhara AA, Elahi R, Johnathan-Lee A, Radulski Ł, et al. Tuberculosis transfronteriza: importación de alpacas infectadas con Mycobacterium bovis del Reino Unido a Polonia y posibilidad de realizar ensayos de diagnóstico serológico en la detección de animales con falsos negativos en la prueba cutánea de la tuberculina. Transbound emerg dis. (2020) 67:1306–14. doi: 10.1111/tbed.13471
Resumen de PubMed | Texto completo de Crossref | Google Académico
8. Kasai H, Ezaki T, Harayama S. Diferenciación de micobacterias de crecimiento lento filogenéticamente relacionadas por sus secuencias gyrB. J Clin Microbiol. (2000) 38:301–8. doi: 10.1128/JCM.38.1.301-308.2000
Resumen de PubMed | Texto completo de Crossref | Google Académico
9. Ghielmetti G, Kupca AM, Hanczaruk M, Friedel U, Weinberger H, Revilla-Fernández S, et al. Infecciones por Mycobacterium microti en ciervos rojos criados en libertad (Cervus elaphus). Emerg Infect Dis. (2021) 27:2025–32. doi: 10.3201/eid2708.210634
Resumen de PubMed | Texto completo de Crossref | Google Académico
10. Michelet L, de Cruz K, Zanella G, Aaziz R, Bulach T, Karoui C, et al. Infección por Mycobacterium microti en animales en Francia. J Clin Microbiol. (2015) 53:981–5. doi: 10.1128/JCM.02713-14
Resumen de PubMed | Texto completo de Crossref | Google Académico
11. Michelet L, de Cruz K, Tambosco J, Hénault S, Boschiroli M-L. Mycobacterium microti interfiere con la vigilancia de la tuberculosis bovina. Microorganismos. (2020) 8:1850. doi: 10.3390/microorganismos8121850
Resumen de PubMed | Texto completo de Crossref | Google Académico
12. Smith NH, Crawshaw T, Parry J, Birtles RJ. Mycobacterium microti: más diverso de lo que se pensaba. J Clin Microbiol. (2009) 47:2551–9. doi: 10.1128/JCM.00638-09
Resumen de PubMed | Texto completo de Crossref | Google Académico
13. Tagliapietra V, Boniotti MB, Mangeli A, Karaman I, Alborali G, Chiari M, et al. Mycobacterium microti en la interfaz entre el medio ambiente y la vida silvestre. Microorganismos. (2021) 9:2084. doi: 10.3390/microorganismos9102084
Resumen de PubMed | Texto completo de Crossref | Google Académico
14. Infantes-Lorenzo JA, Whitehead CE, Moreno I, Bezos J, Roy A, Domínguez L, et al. Desarrollo y evaluación de un ensayo serológico para el diagnóstico de tuberculosis en alpacas y llamas. Frente Vet Sci. (2018) 5:189. doi: 10.3389/fvets.2018.00189
Resumen de PubMed | Texto completo de Crossref | Google Académico
15. Infantes-Lorenzo JA, Dave D, Moreno I, Anderson P, Lesellier S, Gormley E, et al. Nueva plataforma serológica para la detección de anticuerpos contra el complejo Mycobacterium tuberculosis en tejones europeos. Vet Med Sci. (2019) 5:61–9. doi: 10.1002/vms3.134
Resumen de PubMed | Texto completo de Crossref | Google Académico
16. O’Brien A, Clarke J, Hayton A, Adler A, Cutler K, Shaw DJ, et al. Diagnostic accuracy of the Enferplex Bovine Tuberculosis antibody test in cattle sera. Sci Rep. (2023) 13:1875. doi: 10.1038/s41598-023-28410-9
17. Lo Y-T, Shih T-C, Pai T-W, Ho L-P, Wu J-L, Chou H-Y. Conformational epitope matching and prediction based on protein surface spiral features. BMC Genom. (2021) 22:116. doi: 10.1186/s12864-020-07303-5
18. Aranaz A, Liébana E, Mateos A, Dominguez L, Vidal D, Domingo M, et al. Spacer oligonucleotide typing of Mycobacterium bovis strains from cattle and other animals: a tool for studying epidemiology of tuberculosis. J Clin Microbiol. (1996) 34:2734–40. doi: 10.1128/jcm.34.11.2734-2740.1996
19. Kremer K, van Soolingen D, van Embden J, Hughes S, Inwald J, Hewinson G. Mycobacterium microti: more widespread than previously thought. J Clin Microbiol. (1998) 36:2793–4. doi: 10.1128/JCM.36.9.2793-2794.1998
20. Brodin P, Eiglmeier K, Marmiesse M, Billault A, Garnier T, Niemann S, et al. Bacterial artificial chromosome-based comparative genomic analysis identifies Mycobacterium microti as a natural ESAT-6 deletion mutant. Infect Immun. (2002) 70:5568–78. doi: 10.1128/IAI.70.10.5568-5578.2002
21. Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. (2002) 99:3684–9. doi: 10.1073/pnas.052548299
22. Guimaraes AMS, Zimpel CK. Mycobacterium bovis: from genotyping to genome sequencing. Microorganisms. (2020) 8:667. doi: 10.3390/microorganisms8050667
23. Landolt P, Stephan R, Scherrer S. Development of a new High Resolution Melting (HRM) assay for identification and differentiation of Mycobacterium tuberculosis complex samples. Sci Rep. (2019) 9:1850. doi: 10.1038/s41598-018-38243-6
24. De la Fuente J, Diez-Delgado I, Contreras M, Vicente J, Cabezas-Cruz A, Tobes R, et al. La genómica comparativa de aislados de campo de Mycobacterium bovis y M. caprae proporciona evidencia de posibles correlaciones con la viabilidad y virulencia bacteriana. PLoS negl trop dis. (2015) 9:E0004232. doi: 10.1371/journal.pntd.0004232
Resumen de PubMed | Texto completo de Crossref | Google Académico
25. Meehan CJ, Goig GA, Kohl TA, Verboven L, Dippenaar A, Ezewudo M, et al. Secuenciación del genoma completo de Mycobacterium tuberculosis: estándares actuales y cuestiones pendientes. Nat Rev Microbiol. (2019) 17:533–45. doi: 10.1038/s41579-019-0214-5
Resumen de PubMed | Texto completo de Crossref | Google Académico
26. Reis AC, Cunha MV. La estimación de todo el genoma de la recombinación, la mutación y la selección positiva ilumina los impulsores de la diversificación de Mycobacterium bovis. Sci Rep. (2021) 11:18789. DOI: 10.1038/s41598-021-98226-Y
Resumen de PubMed | Texto completo de Crossref | Google Académico
27. Reis AC, Cunha MV. La arquitectura del pangenoma abierto y el paisaje de virulencia de Mycobacterium bovis. Genoma Microb. (2021) 7:000664. doi: 10.1099/mgen.0.000664
Resumen de PubMed | Texto completo de Crossref | Google Académico
28. Lorente-Leal V, Liandris E, Pacciarini M, Botelho A, Kenny K, Loyo B, et al. PCR directa en muestras de tejido para la detección del complejo Mycobacterium tuberculosis: una alternativa al cultivo bacteriológico. J Clin Microbiol. (2021) 59:20. doi: 10.1128/JCM.01404-20
Resumen de PubMed | Texto completo de Crossref | Google Académico
29. Thierry D, Cave MD, Eisenach KD, Crawford JT, Bates JH, Gicquel B, et al. IS6110, un elemento similar al IS del complejo Mycobacterium tuberculosis. Ácidos Nucl Res. (1990) 18:188. doi: 10.1093/nar/18.1.188
Resumen de PubMed | Texto completo de Crossref | Google Académico
30. Kamerbeek J, Schouls L, Kolk A, Van Agterveld M, van Soolingen D, Kuijper S, et al. Detección y diferenciación simultánea de cepas de Mycobacterium tuberculosis para diagnóstico y epidemiología. J Clin Microbiol. (1997) 35:907–14. DOI: 10-1128/JCM.35.4.907-914.1997
Resumen de PubMed | Texto completo de Crossref | Google Académico
31. Bogaerts B, Delcourt T, Soetaert K, Boarbi S, Ceyssens PJ, Winand R, et al. Un flujo de trabajo bioinformático de secuenciación del genoma completo para el análisis clínico de aislados del complejo Mycobacterium tuberculosis, validado utilizando una colección de referencia ampliamente caracterizada con métodos convencionales y enfoques in silico. J Clin Microbiol. (2021) 59:21. doi: 10.1128/JCM.00202-21
Resumen de PubMed | Texto completo de Crossref | Google Académico
32. Bolger AM, Lohse M, Usadel B. Trimmomatic: un recortador flexible para datos de secuencia de iluminación. Bioinformática. (2014) 30:2114–20. doi: 10.1093/bioinformática/btu170
Resumen de PubMed | Texto completo de Crossref | Google Académico
33. Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. Uso de SPAdes de novo assembler. Curr Protocol Bioinformat. (2020) 70:E102. doi: 10.1002/cpbi.102
Resumen de PubMed | Texto completo de Crossref | Google Académico
34. Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: herramienta de evaluación de la calidad para ensamblajes de genomas. Bioinformática. (2013) 29:1072–5. doi: 10.1093/bioinformática/BTT086
Resumen de PubMed | Texto completo de Crossref | Google Académico
35. Wood DE, Lu J, Langmead B. Análisis metagenómico mejorado con Kraken 2. Genoma Biol. (2019) 20:257. doi: 10.1186/s13059-019-1891-0
Resumen de PubMed | Texto completo de Crossref | Google Académico
36. Langmead B, Salzberg SL. Alineación rápida de lectura con pajarita 2. Métodos Nat. (2012) 9:357–9. doi: 10.1038/nmeth.1923
Resumen de PubMed | Texto completo de Crossref | Google Académico
37. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. La alineación/formato de mapa de secuencia y SAMtools. Bioinformática. (2009) 25:2078–9. doi: 10.1093/bioinformática/BTP352
Resumen de PubMed | Texto completo de Crossref | Google Académico
38. van Soolingen D, van der Zanden AGM, de Haas PEW, Noordhoek GT, Kiers A, Foudraine NA, et al. Diagnóstico de infecciones por Mycobacterium microti en humanos mediante el uso de nuevos marcadores genéticos. J Clin Microbiol. (1998) 36:1840–5. doi: 10.1128/JCM.36.7.1840-1845.1998
Resumen de PubMed | Texto completo de Crossref | Google Académico
39. Orgeur M, Frigui W, Pawlik A, Clark S, Williams A, Ates LS, et al. Análisis patogénicos de Mycobacterium microti, un miembro del complejo Mycobacterium tuberculosis con deleción de ESX-1 que causa enfermedad en varios huéspedes. Genoma microbiano. (2021) 7:E000505. doi: 10.1099/mgen.0.000505
Resumen de PubMed | Texto completo de Crossref | Google Académico
40. García-Pelayo MC, Caimi KC, Inwald JK, Hinds J, Bigi F, Romano MI, et al. El análisis de microarrays de Mycobacterium microti revela la deleción de genes que codifican proteínas PE-PPE y antígenos de la familia ESAT-6. Microarrays de tubercul Mycobacterium tuberculosis. (2004) 84:159–66. doi: 10.1016/j.tube.2003.12.002
Resumen de PubMed | Texto completo de Crossref | Google Académico
41. Malm S, Linguissi LSG, Tekwu EM, Vouvoungui JC, Kohl TA, Beckert P, et al. Nuevo sublinaje del complejo Mycobacterium tuberculosis, Brazzaville, Congo. Emerg Infect Dis. (2017) 23:423–9. doi: 10.3201/eid2303.160679
Resumen de PubMed | Texto completo de Crossref | Google Académico
42. Boniotti MB, Gaffuri A, Gelmetti D, Tagliabue S, Chiari M, Mangeli A, et al. Detección y caracterización molecular de aislados de Mycobacterium microti en jabalí del norte de Italia. J Clin Microbiol. (2014) 52:2834–43. doi: 10.1128/JCM.00440-14
Resumen de PubMed | Texto completo de Crossref | Google Académico
43. Brites D, Loiseau C, Menardo F, Borrell S, Boniotti MB, Warren R, et al. Un nuevo marco filogenético para el complejo Mycobacterium tuberculosis adaptado a los animales. Microbiol frontal. (2018) 9:2820. doi: 10.3389/fmicb.2018.02820
Resumen de PubMed | Texto completo de Crossref | Google Académico
44. Bogaerts B, Van den Bossche A, Verhaegen B, Delbrassinne L, Mattheus W, Nouws S, et al. Cerrando la brecha: La secuenciación R10 de Oxford Nanopore Technologies permite obtener resultados comparables a la secuenciación de illumina para la investigación de brotes de patógenos bacterianos basada en SNP. J Clin Microbiol. (2024) 62:E01576–23. doi: 10.1128/jcm.01576-23
Resumen de PubMed | Texto completo de Crossref | Google Académico
45. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Doce años de SAMtools y BCFtools. Gigaciencia. (2021) 10:giab008. doi: 10.1093/gigascience/giab008
Resumen de PubMed | Texto completo de Crossref | Google Académico
46. Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Análisis filogenético rápido de grandes muestras de secuencias de genoma completo bacteriano recombinante utilizando Gubbins. Ácidos nucleicos Res. (2015) 43:E15. doi: 10.1093/nar/gku1196
Resumen de PubMed | Texto completo de Crossref | Google Académico
47. Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: una versión mejor y más rápida de la herramienta de búsqueda de fagos PHAST. Ácidos nucleicos Res. (2016) 44:W16–21. doi: 10.1093/nar/gkw387
Resumen de PubMed | Texto completo de Crossref | Google Académico
48. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: análisis de genética evolutiva molecular en plataformas informáticas. Mol Biol Evol. (2018) 35:1547–9. doi: 10.1093/molbev/msy096
Resumen de PubMed | Texto completo de Crossref | Google Académico
49. Xia E, Teo Y-Y, Ong RT-H. SpoTyping: poligotipado rápido y preciso in silico de Mycobacterium a partir de lecturas de secuencias. Genoma Med. (2016) 8:19. doi: 10.1186/s13073-016-0270-7
Resumen de PubMed | Texto completo de Crossref | Google Académico
50. Cole ST. Genómica comparativa y funcional del complejo Mycobacterium tuberculosis. Microbiología. (2002) 148:2919–28. doi: 10.1099/00221287-148-10-2919
Resumen de PubMed | Texto completo de Crossref | Google Académico
51. Frota CC, Hunt DM, Buxton RS, Rickman L, Hinds J, Kremer K, et al. Estructura del genoma del bacilo Mycobacterium microti, miembro del complejo Mycobacterium tuberculosis con una baja virulencia para los humanos. Microbiología. (2004) 150:1519–27. doi: 10.1099/mic.0.26660-0
Resumen de PubMed | Texto completo de Crossref | Google Académico
52. Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, et al. Compatibilidad variable huésped-patógeno en Mycobacterium tuberculosis. Proc Natl Acad Sci USA. (2006) 103:2869–73. doi: 10.1073/pnas.0511240103
Resumen de PubMed | Texto completo de Crossref | Google Académico
53. Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, et al. Genómica comparativa de vacunas BCG por microarray de ADN del genoma completo. Ciencia. (1999) 284:1520–3. doi: 10.1126/ciencia.284.5419.1520
Resumen de PubMed | Texto completo de Crossref | Google Académico
54. Seemann T. Prokka: anotación rápida del genoma procariota. Bioinformática. (2014) 30:2068–9. doi: 10.1093/bioinformática/btu153
Resumen de PubMed | Texto completo de Crossref | Google Académico
55. Página AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: análisis rápido del genoma de la sartén procariota a gran escala. Bioinformática. (2015) 31:3691–3. doi: 10.1093/bioinformática/BTV421
Resumen de PubMed | Texto completo de Crossref | Google Académico
56. Teufel F, Almagro Armenteros JJ, Johansen AR, Gíslason MH, Pihl SI, Tsirigos KD, et al. SignalP 6.0 predice los cinco tipos de péptidos señal utilizando modelos de lenguaje de proteínas. Nat Biotechnol. (2022) 40:1023–5. doi: 10.1038/s41587-021-01156-3
Resumen de PubMed | Texto completo de Crossref | Google Académico
57. Bendtsen JD, Kiemer L, Fausbøll A, Brunak S. Secreción de proteínas no clásicas en bacterias. BMC Microbiol. (2005) 5:58. doi: 10.1186/1471-2180-5-58
Resumen de PubMed | Texto completo de Crossref | Google Académico
58. Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, et al. PSORTb 3.0: predicción mejorada de la localización subcelular de proteínas con subcategorías de localización refinadas y capacidades predictivas para todos los procariotas. Bioinformática. (2010) 26:1608–15. doi: 10.1093/bioinformática/BTQ249
Resumen de PubMed | Texto completo de Crossref | Google Académico
59. Shen H-B, Chou K-C. Gpos-mPLoc: un enfoque de arriba hacia abajo para mejorar la calidad de la predicción de la localización subcelular de proteínas bacterianas grampositivas. PPL. (2009) 16:1478–84. doi: 10.2174/092986609789839322
Resumen de PubMed | Texto completo de Crossref | Google Académico
60. Yu C-S, Chen Y-C, Lu C-H, Hwang J-K. Predicción de la localización subcelular de proteínas. Proteínas. (2006) 64:643–51. doi: 10.1002/prot.21018
Resumen de PubMed | Texto completo de Crossref | Google Académico
61. Biet F, Boschiroli ML. Infecciones micobacterianas no tuberculosas de relevancia veterinaria. Res Vet Sci. (2014) 97:S69-77. doi: 10.1016/j.rvsc.2014.08.007
Resumen de PubMed | Texto completo de Crossref | Google Académico
62. Infantes-Lorenzo JA, Moreno I, Risalde MÁ, Roy Á, Villar M, Romero B, et al. Caracterización proteómica de derivados proteicos purificados bovinos y aviares e identificación de antígenos específicos para el serodiagnóstico de la tuberculosis bovina. Clin Proteom. (2017) 14:36. doi: 10.1186/s12014-017-9171-z
Resumen de PubMed | Texto completo de Crossref | Google Académico
63. Varela-Castro L, Barral M, Arnal MC, Fernández de Luco D, Gortázar C, Garrido JM, et al. Más allá de la tuberculosis: diversidad e implicaciones de las micobacterias no tuberculosas en la interfaz fauna-ganadería. Transbound emerg dis. (2022) 69:E2978-93. doi: 10.1111/tbed.14649
Resumen de PubMed | Texto completo de Crossref | Google Académico
64. Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, et al. Predicción de alto rendimiento de la antigenicidad de proteínas utilizando datos de microarrays de proteínas. Bioinformática. (2010) 26:2936–43. doi: 10.1093/bioinformática/BTQ551
Resumen de PubMed | Texto completo de Crossref | Google Académico
65. Doytchinova IA, Flower DR. VaxiJen: un servidor para la predicción de antígenos protectores, antígenos tumorales y vacunas de subunidades. BMC Bioinform. (2007) 8:4. doi: 10.1186/1471-2105-8-4
Resumen de PubMed | Texto completo de Crossref | Google Académico
66. Chou PY, Fasman GD. Predicción de la estructura secundaria de las proteínas a partir de su secuencia de aminoácidos. adv enzymol relat areas mol biol. (1978) 47:145–8. doi: 10.1002/9780470122921.ch2
Resumen de PubMed | Texto completo de Crossref | Google Académico
67. Emini EA, Hughes JV, Perlow DS, Boger J. Inducción de un anticuerpo neutralizante del virus de la hepatitis A por un péptido sintético específico del virus. J Virol. (1985) 55:836–9. doi: 10.1128/jvi.55.3.836-839.1985
Resumen de PubMed | Texto completo de Crossref | Google Académico
68. Karplus PA, Schulz GE. Predicción de la flexibilidad de la cadena en proteínas. Naturwissenschaften. (1985) 72:212–3. DOI: 10.1007/BF01195768
69. Parker JMR, Guo D. Nueva escala de hidrofilicidad derivada de datos de retención de péptidos de cromatografía líquida de alta resolución: ¿correlación de los residuos superficiales previstos con la antigenicidad y los sitios accesibles derivados de rayos X? Bioquímica. (1986) 25:5425–32. doi: 10.1021/bi00367a013
Resumen de PubMed | Texto completo de Crossref | Google Académico
70. Kolaskar AS, Tongaonkar PC. Un método semi-empírico para la predicción de determinantes antigénicos en antígenos proteicos. FEBS Lett. (1990) 276:172–4. doi: 10.1016/0014-5793(90)80535-Q
Resumen de PubMed | Texto completo de Crossref | Google Académico
71. Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: mejora de la predicción de epítopos de células B basada en secuencias mediante epítopos conformacionales. Ácidos Nucl Res. (2017) 45:W24–29. doi: 10.1093/nar/gkx346
Resumen de PubMed | Texto completo de Crossref | Google Académico
72. Andersen PH, Nielsen M, Lund O. Predicción de residuos en epítopos discontinuos de células B utilizando estructuras 3D de proteínas. Ciencia de la proteína. (2006) 15:2558–67. doi: 10.1110/ps.062405906
Resumen de PubMed | Texto completo de Crossref | Google Académico
73. Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: una nueva herramienta basada en la estructura para la predicción de epítopos de anticuerpos. BMC Bioinform. (2008) 9:514. doi: 10.1186/1471-2105-9-514
Resumen de PubMed | Texto completo de Crossref | Google Académico
74. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Predicción de la estructura de proteínas de alta precisión con AlphaFold. Naturaleza. (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
Resumen de PubMed | Texto completo de Crossref | Google Académico
75. Barbier M, Wirth T. Historia evolutiva, demografía y propagación del complejo Mycobacterium tuberculosis. Microbiol Spectr. (2016) 4:8. doi: 10.1128/microbiolspec. TBTB2-0008-2016
Resumen de PubMed | Texto completo de Crossref | Google Académico
76. Michelet L, Richomme C, Réveillaud E, Cruz KD, Moyen J-L, Boschiroli ML. Infección por Mycobacterium microti en zorros rojos en Francia. Microorganismos. (2021) 9:1257. doi: 10.3390/microorganismos9061257
Resumen de PubMed | Texto completo de Crossref | Google Académico
77. Pérez de Val B, Sanz A, Soler M, Allepuz A, Michelet L, Boschiroli ML, et al. Infección por Mycobacterium microti en jabalí en libertad, España, 2017-2019. Emerg Infect Dis. (2019) 25:2152–4. doi: 10.3201/eid2511.190746
Resumen de PubMed | Texto completo de Crossref | Google Académico
78. Crispell J, Zadoks RN, Harris SR, Paterson B, Collins DM, de-Lisle GW, et al. Uso de la secuenciación del genoma completo para investigar la transmisión en un sistema de múltiples huéspedes: tuberculosis bovina en Nueva Zelanda. BMC Genom. (2017) 18:180. doi: 10.1186/s12864-017-3569-x
Resumen de PubMed | Texto completo de Crossref | Google Académico
79. Merker M, Kohl TA, Niemann S, Supply P. Evolución de la tipificación de cepas en el complejo Mycobacterium tuberculosis. En:Gagneux S, , editor. Variación de cepas en el complejo Mycobacterium tuberculosis: su papel en biología, epidemiología y control, avances en medicina experimental y biología. Cham: Springer (2017). págs. 43-78.
80. Akhmetova A, Guerrero J, McAdam P, Salvador LCM, Crispell J, Lavery J, et al. Epidemiología genómica de la infección por Mycobacterium bovis en poblaciones simpátricas de tejón y ganado en Irlanda del Norte. Genoma Microb. (2023) 9:mgen001023. doi: 10.1099/mgen.0.001023
Resumen de PubMed | Texto completo de Crossref | Google Académico
81. Duault H, Michelet L, Boschiroli M-L, Durand B, Canini L. Un modelo evolutivo bayesiano para comprender la contribución de la fauna silvestre a la transmisión de Mycobacterium bovis de la familia F4 en el suroeste de Francia. Res. Veterinaria. (2022) 53:28. doi: 10.1186/s13567-022-01044-x
Resumen de PubMed | Texto completo de Crossref | Google Académico
82. Perea C, Ciaravino G, Stuber T, Thacker TC, Robbe-Austerman S, Allepuz A, et al. El análisis de SNP del genoma completo identifica grupos de transmisión putativos de Mycobacterium bovis en el ganado y la fauna silvestre en Cataluña, España. Microorganismos. (2021) 9:1629. doi: 10.3390/microorganismos9081629
Resumen de PubMed | Texto completo de Crossref | Google Académico
83. van Tonder AJ, Thornton MJ, Conlan AJK, Jolley KA, Goolding L, Mitchell AP, et al. Inferencia de la transmisión de Mycobacterium bovis entre ganado y tejones utilizando aislados del Ensayo Aleatorio de Sacrificio de Tejones. PLoS Pathog. (2021) 17:E1010075. doi: 10.1371/journal.ppat.1010075
Resumen de PubMed | Texto completo de Crossref | Google Académico
84. Dippenaar A, Parsons SDC, Sampson SL, van der Merwe RG, Drewe JA, Abdallah AM, et al. Análisis de la secuencia del genoma completo de Mycobacterium suricattae. Tuberculosis. (2015) 95:682–8. doi: 10.1016/j.tube.2015.10.001
Resumen de PubMed | Texto completo de Crossref | Google Académico
85. Cerezo-Cortés M, Rodríguez-Castillo J, Hernández-Pando R, Murcia M. Circulación del genotipo Beijing de M. tuberculosis en América Latina y el Caribe. Pathog Glob Salud. (2019) 113:336–51. doi: 10.1080/20477724.2019.1710066
Resumen de PubMed | Texto completo de Crossref | Google Académico
86. Zheng H, Lu L, Wang B, Pu S, Zhang X, Zhu G, et al. Bases genéticas de la atenuación de la virulencia reveladas por el análisis genómico comparativo de la cepa H37Ra de Mycobacterium tuberculosis frente a la H37Rv. PLoS ONE. (2008) 3:e2375. doi: 10.1371/journal.pone.0002375
Resumen de PubMed | Texto completo de Crossref | Google Académico
87. Stavrum R, Valvatne H, Bø TH, Jonassen I, Hinds J, Butcher PD, et al. Diversidad genómica entre aislados de Mycobacterium tuberculosis de Pekín y de otros países de Myanmar. PLoS UNO. (2008) 3:e1973. doi: 10.1371/journal.pone.0001973
Resumen de PubMed | Texto completo de Crossref | Google Académico
88. Gordon SV, Brosch R, Billault A, Garnier T, Eiglmeier K, Cole ST. Identificación de regiones variables en los genomas de bacilos tuberculosos mediante matrices de cromosomas artificiales bacterianos. Mol Microbiol. (1999) 32:643–55. doi: 10.1046/j.1365-2958.1999.01383.x
Resumen de PubMed | Texto completo de Crossref | Google Académico
89. Gómez-González PJ, Campino S, Phelan JE, Clark TG. Secuenciación portátil de Mycobacterium tuberculosis para aplicaciones clínicas y epidemiológicas. Breve Bioinforme. (2022) 23:BBAC256. doi: 10.1093/bib/bbac256
Resumen de PubMed | Texto completo de Crossref | Google Académico
90. Coscolla M, Gagneux S, Menardo F, Loiseau C, Ruiz P, Borrell S, et al. La filogenómica de Mycobacterium africanum revela un nuevo linaje y una historia evolutiva compleja. Genoma Microbiol. (2021) 7:E000477. doi: 10.1099/mgen.0.000477
Resumen de PubMed | Texto completo de Crossref | Google Académico
91. Tsolaki AG, Gagneux S, Pym AS, Goguet de la Salmoniere YOL, Kreiswirth BN, Van Soolingen D, et al. Las deleciones genómicas clasifican las cepas Beijing/W como un linaje genético distinto de Mycobacterium tuberculosis. J Clin Microbiol. (2005) 43:3185–91. doi: 10.1128/JCM.43.7.3185-3191.2005
Resumen de PubMed | Texto completo de Crossref | Google Académico
92. Sampson SL, Warren RM, Richardson M, Victor TC, Jordaan AM. Polimorfismo de deleción mediado por IS6110 en la región de repetición directa de aislados clínicos de Mycobacterium tuberculosis. J Bacteriol. (2003) 185:2856–66. doi: 10.1128/JB.185.9.2856-2866.2003
Resumen de PubMed | Texto completo de Crossref | Google Académico
93. Al-Khodari NY, Al-Attiyah R, Mustafa AS. Identificación, potencial diagnóstico y expresión natural de péptidos seroreactivos inmunodominantes codificados por cinco regiones genómicas específicas de Mycobacterium tuberculosis. Clin Vac Immunol. (2011) 18:477–82. doi: 10.1128/CVI.00405-10
Resumen de PubMed | Texto completo de Crossref | Google Académico
94. Lyashchenko KP, Grandison A, Keskinen K, Sikar-Gang A, Lambotte P, Esfandiari J, et al. Identificación de nuevos antígenos reconocidos por anticuerpos séricos en tuberculosis bovina. Clin Vac Immunol. (2017) 24:E00259–17. doi: 10.1128/CVI.00259-17
Resumen de PubMed | Texto completo de Crossref | Google Académico
95. Mukherjee P, Dutta M, Datta P, Dasgupta A, Pradhan R, Pradhan M, et al. El antígeno Rv3872 codificado por RD1 de Mycobacterium tuberculosis como candidato potencial para el serodiagnóstico de tuberculosis. Clin Microbiol Infectar. (2007) 13:146–52. doi: 10.1111/j.1469-0691.2006.01660.x
Resumen de PubMed | Texto completo de Crossref | Google Académico
96. Xu J-N, Chen J-P, Chen D-L. Eficacia del serodiagnóstico e inmunogenicidad de la proteína de fusión de Mycobacterium tuberculosis compuesta por la proteína filtrada de cultivo de 10 kilodalton, ESAT-6, y el fragmento de dominio extracelular de PPE68. Clin Vac Immunol. (2012) 19:536–44. doi: 10.1128/CVI.05708-11
Resumen de PubMed | Texto completo de Crossref | Google Académico
97. Ren N, JinLi J, Zhou X, Wang J, Ge P, Khan FA, et al. Identificación de nuevos biomarcadores diagnósticos para Mycobacterium tuberculosis y su potencial aplicación en el serodiagnóstico de la tuberculosis humana. Microb Biotechnol. (2018) 11:893–904. doi: 10.1111/1751-7915.13291
Resumen de PubMed | Texto completo de Crossref | Google Académico
98. D’Souza C, Kishore U, Tsolaki AG. La familia PE-PPE de Mycobacterium tuberculosis: proteínas disfrazadas. Inmunobiología. (2023) 228:152321. doi: 10.1016/j.imbio.2022.152321
Resumen de PubMed | Texto completo de Crossref | Google Académico
99. Karboul A, Mazza A. Eventos frecuentes de recombinación homóloga en familias multigénicas de Mycobacterium tuberculosis PE / PPE: papel potencial en la variabilidad antigénica. J Bacteriol. (2008) 190:7838–46. doi: 10.1128/JB.00827-08
Resumen de PubMed | Texto completo de Crossref | Google Académico
100. Moens C, Filée P, Boes A, Alie C, Dufrasne F, André E, et al. Identificación de nuevos antígenos de Mycobacterium bovis y desarrollo de un inmunoensayo serológico multiplexado para el diagnóstico de la tuberculosis bovina en bovinos. PLoS UNO. (2023) 18:e0292590. doi: 10.1371/journal.pone.0292590
Resumen de PubMed | Texto completo de Crossref | Google Académico
101. Clifford V, He Y, Zufferey C, Connell T, Curtis N. Ensayos de liberación de interferón gamma para monitorear la respuesta al tratamiento de la tuberculosis: una revisión sistemática. Tuberculosis. (2015) 95:639–50. doi: 10.1016/j.tube.2015.07.002
Resumen de PubMed | Texto completo de Crossref | Google Académico
102. Núñez-García J, Downs SH, Parry JE, Abernethy DA, Broughan JM, Cameron AR, et al. Metanálisis de la sensibilidad y especificidad de las pruebas diagnósticas ante-mortem y post-mortem para la tuberculosis bovina en el Reino Unido e Irlanda. Ant: Vet Med. (2018) 153:94–107. doi: 10.1016/j.prevetmed.2017.02.017
Resumen de PubMed | Texto completo de Crossref | Google Académico
103. Greenaway C, Lienhardt C, Adegbola R, Brusasca P, McAdam K, Menzies D. Respuesta humoral a los antígenos de Mycobacterium tuberculosis en pacientes con tuberculosis en Gambia. Int J Tuberc Lung Dis. (2005) 9:1112–9.
104. Rosenkrands I, Aagaard C, Weldingh K, Brock I, Dziegiel MH, Singh M, et al. Identificación de Rv0222 de RD4 como una nueva diana serodiagnóstica para la tuberculosis. Tuberculosis. (2008) 88:335–43. doi: 10.1016/j.tube.2007.12.001
Resumen de PubMed | Texto completo de Crossref | Google Académico
105. Baassi L, Sadki K, Seghrouchni F, Contini S, Cherki W, Nagelkerke N, et al. Evaluación de un test multiantígeno basado en péptidos epítopos de células B para el serodiagnóstico de la tuberculosis pulmonar. Int J Tuberc Lung Dis. (2009) 13:848–54.
106. Sun P, Ju H, Liu Z, Ning Q, Zhang J, Zhao X, et al. Recursos y herramientas bioinformáticas para la predicción de epítopos conformacionales de células B. Computar Métodos Matemáticos Med. (2013) 2013:E943636. doi: 10.1155/2013/943636
Resumen de PubMed | Texto completo de Crossref | Google Académico
107. Zargaran FN, Akya A, Rezaeian S, Ghadiri K, Lorestani RC, Madanchi H, et al. Epítopos de células B de cuatro antígenos de fimbrias de Klebsiella pneumoniae: un estudio integral in silico para el desarrollo de vacunas. Int J Pept Res ther. (2021) 27:875–86. doi: 10.1007/s10989-020-10134-3
Resumen de PubMed | Texto completo de Crossref | Google Académico
108. Potocnakova L, Bhide M, Pulzova LB. Una introducción al mapeo de epítopos de células B y la predicción de epítopos in silico. J Immunol Res. (2016) 2016:E6760830. doi: 10.1155/2016/6760830
Resumen de PubMed | Texto completo de Crossref | Google Académico
109. Roupie V, Alonso-Velasco E, Van Der Heyden S, Holbert S, Duytschaever L, Berthon P, et al. Evaluación de las respuestas de interferón gamma y anticuerpos específicos de micobacterias antes y después de una única prueba cutánea intradérmica en bovinos expuestos de forma natural a M. avium subsp paratuberculosis e infectados experimentalmente con M. bovis. Veterinario Immunol Immunopathol. (2018) 196:35–47. doi: 10.1016/j.vetimm.2017.12.007
Resumen de PubMed | Texto completo de Crossref | Google Académico
110. Aagaard C, Brock I, Olsen A, Ottenhoff THM, Weldingh K, Andersen P. Mapeo de la reactividad inmune hacia Rv2653 y Rv2654: dos nuevos antígenos de baja masa molecular encontrados específicamente en el complejo Mycobacterium tuberculosis. J infectar dis. (2004) 189:812–9. doi: 10.1086/381679
Resumen de PubMed | Texto completo de Crossref | Google Académico
111. Moens C, Saegerman C, Fretin D, Marché S. Evaluación de campo de dos ensayos serológicos comerciales para la detección de la tuberculosis bovina. Res Vet Sci. (2023) 159:125–32. doi: 10.1016/j.rvsc.2023.04.004
Resumen de PubMed | Texto completo de Crossref | Google Académico
Palabras clave: Mycobacterium microti, Mycobacterium bovis, tuberculosis, epítopos de células B, diagnóstico, secuenciación del genoma completo, antígenos
Cita: Moens C, Bogaerts B, Lorente-Leal V, Vanneste K, De Keersmaecker SCJ, Roosens NHC, Mostin L, Fretin D y Marché S (2024) Comparación genómica entre Mycobacterium bovis y Mycobacterium microti y análisis in silico de biomarcadores basados en péptidos para el serodiagnóstico. Frente. Vet. Sci. 11:1446930. doi: 10.3389/fvets.2024.1446930
Recibido: 10 de junio de 2024; Aceptado: 28 de agosto de 2024;
Publicado: 20 de septiembre de 2024.
Editado por:
Lorraine Michelet, Agence Nationale de Sécurité Sanitaire de l’Alimentation, de l’Environnement et du Travail (ANSES), Francia
Revisado por:
Maria Beatrice Boniotti, Instituto Zooprofiláctico Experimental de Lombardía y Emilia Romagna (IZSLER), Italia
Wynand Johan Goosen, Universidad de Stellenbosch, Sudáfrica
Derechos de autor © 2024 Moens, Bogaerts, Lorente-Leal, Vanneste, De Keersmaecker, Roosens, Mostin, Fretin y Marché. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia Creative Commons Atribución (CC BY).
*Correspondencia: Charlotte Moens, charlotte.moens@uclouvain.be
†Estos autores han contribuido igualmente a este trabajo
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente a las de sus organizaciones afiliadas, o las del editor, de los editores y de los revisores. Cualquier producto que puede ser evaluada en este artículo o afirmación que puede ser hecha por su El fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía