
Contribución de las explotaciones a la microbiota en la cadena de valor porcina

Contribución de las explotaciones a la microbiota en la cadena de valor porcina
 Pascal Laforge1,2,3,
Pascal Laforge1,2,3,  Antonio T. Vincent1,2,3,
Antonio T. Vincent1,2,3,  Caroline Duchaine3,4,
Caroline Duchaine3,4,  Perrine Feutry2,
Perrine Feutry2,  Annick Dion-Fortier2,
Annick Dion-Fortier2,  Pier-Luc Plante2, Éric Pouliot5, Sylvain Fournaise5 y
Pier-Luc Plante2, Éric Pouliot5, Sylvain Fournaise5 y  Linda Saucier1,2,3*
Linda Saucier1,2,3*- 1Département des sciences animales, Université Laval, Québec, QC, Canadá
- 2Institut sur la nutrition et les aliments fonctionnels, Université Laval, Québec, QC, Canadá
- 3Centre de recherche en infectiologie porcine et avicole, Faculté de médecine vétérinaire, Université de Montréal, Saint-Hyacinthe, QC, Canadá
- 4Département de biochimie, microbiologie et bio-Informatique, Université Laval, Québec, QC, Canadá
- 5Olymel S.E.C./L.P., Boucherville, QC, Canadá
Introducción: Una comprensión profunda de la ecología microbiana dentro de la cadena de valor porcina es esencial para desarrollar nuevas estrategias para optimizar la calidad microbiológica de los productos porcinos. Hasta donde sabemos, ningún estudio hasta la fecha ha seguido la microbiota a través de la cadena de valor desde los animales de granja vivos hasta los cortes de carne obtenidos para el mercado. El objetivo de este estudio es evaluar cómo la microbiota de los cerdos y su entorno influyen en la composición microbiana de las muestras recogidas a lo largo de la cadena de valor, incluida la planta cárnica y los cortes de carne.
Método y resultados: Los resultados de la secuenciación del ADNr 16S, las concentraciones de ácidos grasos de cadena corta y el análisis metabolómico de heces de cerdo revelaron que la microbiota de dos granjas con diferentes estados sanitarios era distintiva. Los recuentos totales de bacterias mesófilas aeróbicas y enterobacteriaceae de muestras recolectadas en la planta de carne después de los pasos de limpieza y desinfección previos a la operación estuvieron en o alrededor del límite de detección y los cerdos de las granjas seleccionadas fueron los primeros en ser sacrificados en cada día de envío. Los recuentos bacterianos de muestras individuales recolectadas en la planta de carne no variaron significativamente entre las granjas. Los resultados de la diversidad alfa indican que a medida que avanzamos en los pasos de la cadena de valor, hay una clara reducción de la diversidad de la microbiota. Un análisis de diversidad beta reveló una microbiota más distinta en las granjas en comparación con la planta de carne, que cambia y se vuelve más uniforme a medida que se toman muestras hacia el final de la cadena de valor. El análisis del rastreador de fuentes mostró que solo el 12,92% de la microbiota en las muestras de hombro se originó en las granjas y el 81% de las bacterias detectadas en los cadáveres vestidos eran de origen desconocido.
Discusión: En general, los resultados sugieren que con el nivel actual de control microbiano en las granjas, es posible obtener productos de cerdo con calidad microbiológica similar de diferentes granjas. Sin embargo, se requieren estudios más amplios para determinar el impacto del estado sanitario del rebaño en los productos finales.
1 Introducción
La carne de cerdo constituye una parte importante del consumo mundial de carne y, hasta 2015, era la carne más consumida del mundo (OCDE, 2021). Como todos los músculos de animales sanos, con la excepción de los ganglios linfáticos, la carne de cerdo alberga un pequeño número de microorganismos (Huffman, 2002). La carne y los productos cárnicos a menudo se identifican como causantes de enfermedades transmitidas por los alimentos (Painter et al., 2013; Ramsay y Delisle, 2017). La carne es rica en nutrientes y agua, lo que apoya el crecimiento microbiano (Remenant et al., 2015; Zagorec y Champomier-Vergès, 2017). Como tal, la contaminación de las canales de cerdos y los cortes de carne resultantes pueden provocar deterioro o crecimiento de patógenos a lo largo de la vida útil del producto. La contaminación de la carne puede ocurrir a través de microorganismos que están presentes en diferentes partes del animal (tracto digestivo, piel, tracto respiratorio, saliva, etc.; Zweifel y Stephan, 2014). Por lo tanto, la calidad de la carne de cerdo se basa en una combinación de medidas efectivas de bioseguridad, gestión de la salud del rebaño en la granja, prácticas higiénicas de sacrificio y corte y medidas de gestión de riesgos en toda la cadena de valor (planes HACCP y medidas similares).
El estado sanitario o estado de salud de una granja es determinado por un veterinario a través del monitoreo de enfermedades actuales e históricas, así como las condiciones sanitarias actuales de la granja. Se cree que este estado influye en la calidad microbiana final del producto (Hurd et al., 2008; Vigors et al., 2020). La gestión sanitaria adecuada y otras medidas sanitarias, incluida la bioseguridad en la explotación, reducen el riesgo de infección por los principales patógenos porcinos, como los serovares patógenos de Mycoplasma hyopneumoniae (neumonía enzoótica) y Actinobacillus pleuropneumoniae (pleuroneumonía; Stärk y otros, 2008). Otros patógenos, incluyendo Clostridium perfringens, Salmonella enterica y Yersinia enterocolitica se han relacionado con la contaminación de cadáveres (Fosse et al., 2008). Si bien las granjas porcinas modernas generalmente mantienen un excelente estado sanitario, existe un cierto nivel de variabilidad entre las granjas (Cameron, 2000). Mejorar las condiciones sanitarias en la granja también puede mejorar la salud y el comportamiento de los animales (Meer et al., 2017). Para evaluar adecuadamente el impacto del estado sanitario de las explotaciones en la microbiota a lo largo de las diferentes etapas de la cadena de valor, se debe obtener información precisa sobre las poblaciones microbianas en cerdos, desde la granja hasta el envasado de la carne. Hasta la fecha, la única información disponible se limita a los estudios en los que las muestras se tomaron aleatoria o esporádicamente en etapas específicas de la cadena de valor.
Varios estudios de secuenciación de amplicones de ARNr 16S se han centrado en áreas clave de la cadena de valor porcina. El ecosistema microbiano encontrado en el aire del edificio de viviendas (Nehme et al., 2008; Kumari et al., 2016; Kraemer et al., 2019; Yan et al., 2021), el intestino porcino (Kim et al., 2015; Kim e Isaacson, 2015; Zhao et al., 2015; Holman et al., 2017; Crespo-Piazuelo et al., 2018; Quan y otros, 2018; Xiao et al., 2018; Adhikari et al., 2019; Mark Welch et al., 2019; Vigors et al., 2020), las vías respiratorias, la boca y la saliva (Wang et al., 2018; Huang et al., 2019; Mou et al., 2019; Murase et al., 2019), el entorno de las plantas cárnicas (Bridier et al., 2019; Zwirzitz et al., 2020) y el cadáver (Mann et al., 2016; Zwirzitz et al., 2019; Zwirzitz et al., 2020) han sido examinados. La microbiota intestinal porcina es, con mucho, el más estudiado de estos entornos, lo suficiente como para que el concepto de una microbiota «central» (bacterias presentes en más del 90% de las muestras) fuera propuesto por Holman et al. (2017). Estos autores identificaron los géneros Clostridium, Blautia, Lactobacillus, Prevotella, Ruminococcus, Roseburia, RC9 «gut group» y Subdoligranulum como los principales miembros de ese «núcleo».
Hasta donde sabemos, ningún otro estudio cubre todos estos entornos y sigue a los mismos animales vivos desde la granja hasta los cortes de carne. Se seleccionaron dos granjas porcinas comerciales con diferentes estados sanitarios, caracterizamos la microbiota de los animales y sus ambientes y observamos cómo cambiaba la microbiota en los pasos de aderezo de la canal (sacrificio, evisceración) y preparación del corte de carne. Este estudio descriptivo presenta un conocimiento detallado de la microbiota de granja y animal, y la eficacia con la que las plantas procesadoras de carne mitigan el impacto de estos microbios en las canales y cortes de carne resultantes.
2 Material y métodos
2.1 Granjas seleccionadas
Dos granjas comerciales de 126 fueron seleccionadas por veterinarios experimentados y estaban ubicadas en la provincia de Quebec. Las granjas fueron seleccionadas en función de su estado sanitario, la salud gastrointestinal de los animales y el historial médico asociado con las granjas; una granja con un estatus sanitario más bajo (granja-L) y otra con un estatus más alto (granja-H). Ambas eran granjas de acabado de tamaño de alojamiento similar, con 1.200 y 1.600 animales, respectivamente. Los animales (hembra (Yorkshire X Landrace) X Duroc macho) fueron alimentados con la misma dieta comercial y siguieron el mismo programa de nutrición de acabado de 4 fases. Hubo una excepción en la granja-L, donde la dieta cambió de pellets a puré de alimento 22 días antes del primer muestreo, debido a un episodio de salmonelosis (ver discusión para detalles clínicos). Los animales de ambas granjas fueron enviados a la misma planta de carne inspeccionada por el gobierno federal cuando alcanzaron un peso de 115 ± 7 kg. Provenían del mismo lote de producción donde se siguieron tres de cada cinco envíos. Las muestras se recogieron en cada granja 3 días antes de que los cerdos fueran transportados al matadero; Los animales de granja L se enviaron de julio a agosto y la granja H de octubre a noviembre. Los animales estaban sujetos a una retirada obligatoria de alimento de 18 horas antes del sacrificio, tal como se exige en el acuerdo de mercado sobre cuestiones de bienestar animal. Todos los procedimientos de cuidado y manejo de animales fueron aprobados por el Comité de Uso y Cuidado de Animales de la Universidad Laval (2019-329), que se adhiere estrictamente a las Directrices del Consejo Canadiense de Cuidado de Animales (CCAC, 2009).
2.2 Sacrificio y rotura de canales
Los cerdos fueron separados en grupos de cinco y aturdidos usando CO2. Después del aderezo, las canales se enfriaron rápidamente durante 90 minutos y luego se enfriaron durante la noche (24 h, 2 ° C) antes de dividirse en cortes minoristas (Figura suplementaria S1). Se recogieron los datos de inspección, incluida la frecuencia de los deméritos (absceso, linfadenitis, hematomas, etc.) y el número de carasses que fueron condenados o retenidos para un examen adicional (animal muerto en la recepción, abscesos grandes y generalizados, peritonitis, ictericia, etc.), tamaño del estómago (en términos de eficacia de retirada de alimento) y presencia de vísceras hinchadas.
2.3 Muestreo y procesamiento de muestras
Se recogieron múltiples tipos de muestras a lo largo de la cadena de valor (Figura complementaria S1). Todas las muestras se almacenaron en hielo hasta que se procesaron. En las granjas, los tipos de muestra incluían aire (Ar), heces (Fc), saliva (Sa) y alimento (Fe). Se recogieron muestras Fc y Sa de 16 plumas para los tres envíos. Se tomaron muestras de aire utilizando un muestreador de aire seco SASS 3100 (Research International, Monroe, WA, Estados Unidos) con un cartucho de filtro electret estándar soldado. Un volumen de 10 m3 se recolectó a 300 mL/min durante 33 min, tres veces al día (8:00, 11:00 y 14:00). El aparato se colocó sobre una mesa (1 m de altura) en medio del callejón en la habitación donde se alojaban los cerdos. Luego, los filtros se conservaron a -20 ° C hasta que se realizaron extracciones de partículas utilizando el extractor de partículas SASS 3010 (Research International) con el tampón de recuperación (138 mM NaCl, 2.7 mM KCl, 0.05% Triton X-100, <0.1% NaN3 10 mM Na3PO4, pH 7.4). Las extracciones se realizaron de acuerdo con las instrucciones del fabricante. Con el fin de garantizar cantidades suficientes de material para la posterior purificación del ADN, las soluciones de partículas se agruparon en volúmenes iguales según la fecha de muestreo y luego se centrifugaron a 4 °C, 14 000 × g durante 20 min (centrífuga Sorvall legend XTR, Thermo Fisher Scientific, Waltham, MA, Estados Unidos). Se eliminó el exceso de sobrenadante y las células se almacenaron a -80 ° C hasta la extracción del ADN.
Las muestras de Fc se recogieron utilizando un PERFORMAbiome•GUT | Kit de muestreo de PB-200 siguiendo las instrucciones del fabricante (DNAgenotek, Ottawa, Ontario, Canadá). Se recolectaron heces recién defecadas de 16 animales al azar, una por corral, cada día de muestreo. Se recogieron muestras Fc adicionales para análisis metabolómicos y de cadena corta/ácidos grasos volátiles (ver secciones a continuación). Una alícuota de 500 μL de cada tubo de PB-200 se agrupó asépticamente según la fecha de muestreo. Las muestras individuales y agrupadas se almacenaron a -80 ° C hasta la extracción de ADN. La saliva se recolectó utilizando un kit P-151 (DNAgenotek) según lo sugerido por el fabricante. Una alícuota de 250 μL de cada tubo P-151 se agrupó por fecha de muestreo y luego se almacenó a -80 ° C hasta la extracción de ADN. El alimento se muestreó asépticamente directamente de ocho comederos y se almacenó en bolsas Whirlpack estériles de 4 oz (Nasco, Madison, WI, Estados Unidos). Las muestras se agruparon para formar un compuesto mezclando 2,5 g de alimento de cada alimentador y luego se almacenaron a -20 ° C hasta la extracción de ADN.
En la planta de carne, se realizaron procedimientos preoperacionales para garantizar una línea de procesamiento limpia. En cada día de transformación, los animales estudiados fueron los primeros en ser sacrificados y sus canales fueron los primeros en dividirse en cortes de venta al por menor al día siguiente. Esto nos permitió evaluar adecuadamente qué contaminación se originó en los animales de cada granja. Se recogieron muestras ambientales para cada envío antes y después de que los animales en estudio se procesaran en la línea de aderezo. Las muestras de superficie se obtuvieron utilizando una esponja estéril (Whirl-Pak Speci-Sponge Environmental Surface Sampling Bags, Nasco) que se humidificó con 10 ml de agua estéril tamponada al 2% (Peptone Water, tamponada con fosfato, Milipore Sigma, Oakville, Canadá) y una plantilla estéril de 10 × 10 cm (plantilla de ganado de 3 M, USDA100, 3 M Canadá, Londres, Malasia). En colaboración con el equipo de control de calidad, seleccionamos los sitios de muestreo de acuerdo con su plan HACCP. En la evisceración, se recogieron muestras de agua de un drenaje central debajo del transportador de vísceras (Dev; 150 mL). Además, muestras superficiales de un canalón post-evisceración (Gev; 300 cm®®2) y de un transportador antes del primer lavado de la ducha de la canal (Cev; 300 cm2). Veinticinco canales refrigeradas por chorro fueron muestreadas usando hisopos superficiales (Dc). Los hisopos se recolectaron de un 100 cm2 superficie de la pata trasera cerca del ano, el vientre y la papada (300 cm2 en total), de acuerdo con las Directrices para las pruebas de Escherichia coli para la verificación del control de procesos en establecimientos de sacrificio de ganado bovino y porcino (FSIS-GD-1996-0001, 2005, FDA). Un segundo grupo de 25 canales se muestreó al día siguiente después de la refrigeración nocturna a 2 ° C (Cc). En el área donde se dividieron las canales, se recogieron muestras de agua de un drenaje central (Dcu; 150 mL) y muestras de superficie al final del transportador (Ccu; 300 cm2). Durante esta parte del proceso, se seleccionaron aleatoriamente 25 hombros (S) para el muestreo (Figura Suplementaria S1) y un total de 450 cm2 se frotó de la superficie de cada hombro y de la sección interior donde se extrajo el hueso del hombro.
En el laboratorio, se agregaron 10 ml de agua tamponada al 2% a cada una de las bolsas Whirl-Pak que contenían la esponja. El contenido de la bolsa se homogeneizó utilizando un Stomacher 400C (Seward Laboratory Systems Inc., Londres, Reino Unido) durante 2 min, a 230 rpm. Se agrupó una alícuota de 2 ml de cada conjunto de canales y muestras de hombro y se mezcló completamente para la enumeración microbiana. Se consideró necesario agrupar algunas muestras a fin de obtener suficiente ADN para aplicaciones posteriores. Las muestras se almacenaron a -80 ° C hasta la extracción del ADN.
2.4 Análisis de ácidos grasos de cadena corta de heces
Se midieron las concentraciones de ácidos grasos de cadena corta (AGCC; ácidos acético, propiónico, butírico, isobutírico, valérico e isovalérico) en las 16 muestras individuales de Fc de cada día de muestreo (96 muestras en total). Las muestras de Fc se almacenaron a -80 ° C hasta el análisis. Luego se prepararon suspensiones fecales a partir de muestras de Fc descongeladas. Las muestras se dividieron en 500 mg de alícuotas de heces que se disolvieron en 10 veces el volumen de agua y se homogeneizaron durante 2 min con un Bead Ruptor 12 (Omni international, Kennesaw, GA, Estados Unidos). Las suspensiones se centrifugaron a 4 ° C, 5.500 × g durante 30 min y los AGCC se extrajeron del sobrenadante mediante extracción líquido-líquido y se analizaron mediante cromatografía de gases acoplada a un detector de ionización de llama (GC-FID Shimadzu, Kyoto, Japón), como se describe en Roussel et al. (2022).
2.5 Análisis metabolómico de heces
También se realizó metabolómica no dirigida en las muestras individuales de Fc utilizando una cromatografía líquida acoplada a un espectrómetro de masas (LC-MS). Las heces se descongelaron y se dividieron en alícuotas de 900 mg. Las muestras se liofilizaron con un Lyovapor L-300 (BÜCHI Labortechnik AG, Flawil, Suisse) durante 72 h. Se añadió un volumen de 12,5 μL de MeOH al 50% en agua por mg de materia seca. Las muestras se mezclaron con un batidor de cuentas (Bead ruptor 12) durante 2 min y luego se agitaron con un vórtice multitubo (Vx-2500, VWR international, Wayne, NJ, USA) durante 5 min. A continuación, las muestras se sonicaron en un baño ultrasónico a 25 ° C durante 30 min, se agitaron con un vórtice multitubo durante 5 min y se centrifugaron a 4 ° C, 2 795 × g durante 30 min. Se recogió un volumen de 250 μL de sobrenadante y se mezcló con 250 μL de MeOH al 50% en agua que contenía 10 patrones internos (2 ppm de ácido trihidroxibenzoico-d2, 2 ppm de cafeína-metil-d3, 10 ppm de ácido succínico-d6, 0.2 ppm N-dodecilfosfocolina-d38, 10 ppm de ácido transcinámico-d5, 2 ppm L-triptófano-d5, 2 ppm de ácido glicocólico-d4, 10 ppm L-leucina-d7, 2 ppm de ácido 4-hidroxibenzoico-d4 y 2 ppm de metil 4-hidroxibenzoato-D4; Isótopo CDN). Finalmente, las muestras se filtraron con filtros de centrifugado (InnoSep Spin, NY, 0.2 μm, Canadian Life Science, Peterborough, Canadá) antes del análisis LC-MS.
Los análisis LC-MS se realizaron en un sistema que consiste en una cromatografía líquida de ultra alta resolución (UHPLC) Vanquish y un espectrómetro de masas Fusion Tribrid (Thermo Fisher Scientific, Waltham, MA, Estados Unidos). Las fases móviles fueron agua (A) y acetonitrilo (B), cada una con 0,1% de ácido fórmico. Se utilizó el siguiente gradiente de elución: 0 min 2% de B, 0,5 min 2% de B, 9 min 45% de B, 9,5 min 80% de B, 15,5 min 80% de B, 16 min 2% de B y 22 min 2% de B, a un caudal de 0,3 mL/min. Se inyectó un volumen de 5 μL en una columna Acquity UPLC HSS T3 (100 Å, 1,8 μm, 2,1 mm × 100 mm; Waters, Milford, MA, Estados Unidos) y mantenido a 30°C. Las muestras se mantuvieron a 4 °C en el muestreador automático. Se analizó una muestra agrupada de Control de Calidad (QC) que consiste en un volumen igual de cada muestra cada 10 muestras.
Se utilizó un electronebulizador calentado a 350°C como fuente de ionización con una tensión capilar de 3,5 kV en modo positivo y 2,5 kV en modo negativo. Las adquisiciones de espectroscopía de masas (MS) se realizaron en un orbitrap a una resolución de 120 000 en modo de perfil utilizando Easy-IC para la corrección de masa. Todos los demás parámetros se establecieron en sus valores predeterminados. SRA.2 los espectros para la muestra agrupada de control de calidad se adquirieron utilizando el método AcquireX. Los datos se analizaron con los paquetes de software Compound Discovered 3.2 y MetFrag Web 2.1 (Ruttkies et al., 2016).
2.6 Análisis microbiano
Para la enumeración en placas de agar, se realizaron diluciones seriadas diez veces mayores utilizando agua estéril al 0,1% de peptona (BD Biosciences, Franklin Lakes, NJ, Estados Unidos). Los recuentos mesófilos aeróbicos totales (TAM) (Health Canada, 2020) se realizaron en agar de recuento de placas (BD Biosciences; 35 ° C durante 48 h). Los recuentos de enterobacterias (EB) (Health Canada, 1997) se realizaron en agar de bilis y glucosa rojo violeta (BD Biosciences; 35°C durante 24 h). Las mediciones se realizaron por duplicado. Para las muestras ambientales (desagües, transportador y canalón), la contaminación originada en los animales se midió restando el valor obtenido después de los procedimientos previos a la operación del valor obtenido justo después del paso de los animales en estudio. El número de animales enviados a la planta cárnica varió a lo largo de los seis envíos, por lo que se utilizó una ponderación estadística básica para ajustar los resultados a un tamaño de lote de 300 animales (Kalton y Flores-Cervantes, 2003). Todos los recuentos bacterianos se transformaron en un registro10 value of colony forming units per 300 cm2 or 300 mL prior to statistical analysis (Gill, 2000). When no colonies were observed, the level of detection was used in statistical analyses.
2.7 Extracción de ADN y secuenciación de ADNr 16S
Las muestras congeladas se descongelaron a 4°C. Las muestras líquidas se centrifugaron (leyenda de Sorvall) a 4°C, 24 000 × g durante 10 min para las muestras de 50 mL, y 14 000 × g para 20 min para las muestras de 10 mL. Se eliminó el sobrenadante y se utilizaron los gránulos celulares para la extracción de ADN. Las muestras de fe se hidrataron en agua de peptona tamponada al 2% en una proporción de 9:1 de agua para alimentar durante 30 minutos a 4 °C en bolsas estomacales filtradas. Los pellets hidratados se homogeneizaron utilizando un Stomacher 400C durante 2 min a 230 rpm. Se recuperó un volumen total de 2,5 mL y se centrifugó a 14 000 × g durante 10 min a temperatura ambiente. Las células peletizadas se utilizaron para la extracción de ADN.
Los kits de extracción de ADN se eligieron en función de su eficiencia de extracción con las diversas muestras recolectadas. Se utilizó un kit DNeasy PowerSoil (QIAGEN, Toronto, Canadá) para muestras de aire, alimentación, transportador, canalón y carcasa. Se utilizó un QIAamp Fast DNA Stool Mini Kit (QIAGEN) para las heces estabilizadas, el DNeasy PowerWater Kit (QIAGEN) para el líquido recogido de los drenajes y un QIAamp BiOstic Bacteremia DNA Kit (QIAGEN) para las muestras S. Se utilizó un kit completo de purificación de ADN y ARN MasterPure (Lucigen, Teddington, Reino Unido) para muestras de Sa. Todos los kits se utilizaron siguiendo los protocolos del fabricante. Las muestras de ADN extraídas se cuantificaron utilizando un espectrofotómetro NanoDrop One (Thermo Fisher Scientific, Ottawa, ON, Canadá).
2.8 Amplificación del ADN 16S
Durante la configuración preliminar del experimento, se realizó una secuenciación metagenómica de escopeta para analizar las comunidades microbianas. Desafortunadamente, el ADN de cerdo estaba en tal abundancia (>95%) en las muestras recolectadas que el análisis estaba perdiendo profundidad. Por lo tanto, la amplificación y secuenciación del gen 16S se consideró más apropiada. La preparación y secuenciación de la biblioteca se realizaron en el Plateforme d’analyse génomique (Institut de Biologie Intégrative et des Systèmes, Université Laval, Quebec City, Canadá). La amplificación de la región 16S V3-V4 se realizó como se describe en Klindworth et al. (2012) en un enfoque de PCR oligo largo. Las reacciones de PCR se purificaron utilizando un kit de limpieza de PCR Axygen (Axygen, Nueva York, NY, EUA). La calidad del producto de PCR purificado se confirmó con un chip BioAnalyzer DNA7500 (Agilent, Santa Clara, CA, Estados Unidos) y se cuantificó utilizando un espectrofotómetro NanoDrop One. Los amplicones con código de barras se agruparon en concentraciones equimolares y se secuenciaron en un Illumina MiSeq (extremo pareado 300 pb con dos lecturas de índice). Las muestras recogidas después de los procedimientos de limpieza y desinfección preoperatorios con una cantidad de lectura insuficiente se descartaron.
2.9 Procesamiento de secuencias
El primer paso en el procesamiento de lectura fue inspeccionar los gráficos de calidad generados por FastQC (versión 0.11.5; Andrews, 2010). Las variantes de secuencia de amplicón (ASV) se generaron utilizando el paquete de flujo de trabajo DADA2 (versión 1.22.0; R versión 4.1.1; Callahan et al., 2016). Durante la filtración, los primeros 17 nucleótidos de las lecturas directas y las primeras 21 de las lecturas inversas se recortaron para eliminar los cebadores. Las secuencias que contenían nucleótidos ambiguos (N) fueron descartadas. Según los hallazgos de Prodan et al. (2020), el filtro de error esperado no se utilizó para evitar el sesgo hacia las bacterias con un genoma propenso a errores. La desreplicación, la inferencia de muestras, la identificación de quimeras y la fusión de las lecturas de extremo emparejado se realizaron utilizando los parámetros predeterminados, con la excepción de que las muestras se agruparon durante el paso de inferencia. La asignación taxonómica se realizó utilizando la base de datos SILVA rRNA (versión 138.1; Pruesse et al., 2007) con el método de clasificador bayesiano ingenuo (el comando assignTaxonomy del paquete DADA2). A continuación, se añadieron especies con la función añadir especies. Se construyó un árbol filogenético basado en una alineación múltiple (DECIPHER R package version 2.22.0; Wright, 2016). Luego se construyó un árbol de unión vecina y se usó como base para el árbol de máxima verosimilitud GTR + G + I (phangorn, versión 2.8.1 del paquete R; Schliep, 2010). Los recuentos, taxones, metadatos de estudio y árboles filogénicos se combinaron en un objeto phyloseq (versión 1.38.0; McMurdie y Holmes, 2013). Los contaminantes se eliminaron utilizando muestras en blanco y el método de prevalencia (umbral de 0,5; decontam, versión del paquete R 1.14.0; Davis et al., 2018). Los datos se filtraron eliminando los ASV no bacterianos (Reino Eukaryota y Archaea, Orden Cloroplasto y Familia Mitocondria), luego ASV sin identificación de filo seguido de cualquier filo de baja prevalencia (menos de cinco ASV por filo) y, finalmente, ASV de baja prevalencia (presente en menos del 5% de las muestras). Se buscaron ASV filtrados en la base de datos curada NCBI nr/nt 16S (Bioproject 33175 o 33317; excluyendo archea; descargado el 14-01-2022) en GenBank usando BLASTN (versión 2.12.0; Altschul y otros, 1990). Cuando la consulta ASV tenía más del 97% de identidad con las secuencias en la base de datos GenBank, la misma identificación a nivel de género que en la base de datos SILVA y una asignación de especies claramente definida (sin identidad ambigua del mismo porcentaje pero identificación de especies diferente; se dejaron como NA) se reasignaron manualmente a ese ASV. El mismo procedimiento se siguió para la reasignación de género con un umbral de identidad del 90% y consenso familiar. Los taxones restantes se agruparon en la fila «taxones restantes». Los recuentos de ASV se normalizaron en abundancia relativa para la visualización del mapa de calor (ampvis2, versión del paquete R 2.7.13; Andersen et al., 2018). Se produjo un gráfico de mapa de calor utilizando un subconjunto de familias bacterianas conocidas por afectar a la carne (Campylobacteraceae, Carnobacteriaceae, Enterobacteriaceae, Lactobacillaceae y Staphylococcaceae; Baer et al., 2013; Saucier, 2016; Møretrø y Langsrud, 2017). Las secuencias se pueden encontrar en DDBJ/ENA/GenBank bajo el BioProject PRJNA923296.
2.10 Análisis estadístico
Se utilizó una prueba de Shapiro-Wilk para confirmar la normalidad y una prueba T de Student para comparar cada tipo de muestra entre granjas y para los recuentos de TAM y Enterobacteriaceae. También se utilizaron pruebas T para las muestras recogidas antes e inmediatamente después del procesamiento de los animales en estudio, así como para los datos de inspección de canales recogidos para cada envío y los CCPA en las heces. Estas pruebas también se realizaron sin separarlos por granja para determinar si la presencia del animal tenía un efecto sobre el nivel de contaminación. También se realizó un análisis no paramétrico sobre la base de que cuando se utiliza un umbral de concentración microbiana específico para determinar el final de la vida útil, los recuentos celulares más cercanos a ese límite umbral representan un mayor riesgo para la seguridad alimentaria microbiana. Para eso, se realizó una prueba de Wilcoxon para comparar las dos granjas en lugar de una prueba T de Student porque las distribuciones no eran normales cuando todos los tipos de muestra se agruparon.
La integridad de los datos metabolómicos se validó por primera vez midiendo el %CV en diferentes estándares internos detectados en positivo (cafeína-d3, dodecilfosfoclolina-d38, triptófano-d5 y leucina-d7) y negativos (ácido succínico-d6, ácido transcinámico-d5 y triptófano-D5) ionización. El %CV fue inferior al 20% para todos estos compuestos. Luego, la calidad de los datos metabolómicos no dirigidos se evaluó mediante un análisis de componentes principales (ACP) y mostró una fuerte agrupación de inyecciones de QC-pool. Una muestra se consideró un valor atípico y se eliminó. Las características metabólicas se filtraron en función de los siguientes criterios estrictos: presente en todas las inyecciones de QC pool (3183 y 542 características dejadas en modo positivo y negativo), y %CV < 20% en su propio grupo (granja-L o granja-H, 251 y 71 características restantes en cada modo de ionización). Características sin MS2 Los espectros también se eliminaron del análisis (quedan 240 y 64 características). Un análisis diferencial (Log2 Se realizó un cambio de pliegue >1 y un valor p <0,05) en las características restantes. Los metabolitos seleccionados se identificaron supuestamente utilizando la puntuación FiSH de Compound Discoverer como métrica de puntuación y luego se confirmaron utilizando MetFrag Web.
Las secuencias de ADNr 16S se clasificaron según la granja, el tipo de muestra (Ar, Fc, Sa, Fe, Dev, Cev, Gev, Dc, Cc, Dcu, Ccu y S), la ubicación del muestreo (granja, evisceración, recorte) y la semana de envío (semanas 1, 2 y 3). También se realizaron análisis de abundancia diferencial para un subconjunto de géneros y familias importantes para la seguridad de la carne (Campylobacteraceae, Carnobacteriaceae, Enterobacteriaceae, Lactobacillaceae y Staphylococcaceae), entre granjas, utilizando la metodología Análisis de composiciones de microbiomas con corrección de sesgo (ANCOMBC). Este método se realizó en los recuentos de lectura sin procesar de cada familia y su género (paquete ANCOMBC R versión 1.4.0; Lin y Peddada, 2020). Se utilizaron varios parámetros para controlar la tasa de falso descubrimiento (FDR) y aumentar los robusnes de los análisis. Estos incluyeron: un corte cero de 1 (sin géneros excluidos), 1.000 iteraciones, una estimación de varianza conservadora, ajuste FDR de los valores de p, ceros estructurales y ceros negativos de límite inferior.
La diversidad alfa (dentro de la muestra) se calculó sobre datos no normalizados (phyloseq versión 1.30.0; McMurdie y Holmes, 2013). La riqueza de especies se evaluó con un índice Observado y Chao1 y la uniformidad se evaluó con el índice de Shannon y Simpson. Estos índices se seleccionaron para evaluar grandes cambios en la población, ya que dan un número para el ASV total (Observado y Chao1) y la distribución de abundancia entre estos ASV (Shannon y Simpson). Se utilizó una prueba T de Student para comparar cada tipo de muestra entre granjas. Se utilizó una corrección FDR para controlar los falsos positivos. Para evaluar las diferencias entre la diversidad de tipos de muestra, se realizó una prueba Tukey HSD entre todas las muestras (agricolae, versión 1.3-5 del paquete R; Mendiburu y Yaseen, 2021).
La diversidad beta (entre muestras) se calculó para los recuentos normalizados de ASV utilizando distancias UniFrac no ponderadas y ponderadas (Lozupone y Knight, 2005) y diferencias de Bray-Curtis (paquete Phyloseq R versión 1.38.0; McMurdie y Holmes, 2013). Los datos se normalizaron realizando una transformación de Hellinger (decostand function of the vegan R package version 2.5–7; Legendre y Gallagher, 2001). Se utilizó el análisis de coordenadas principales (PCoA) para visualizar las distancias entre muestras (Ampvis2, paquete R versión 2.7.13; Andersen et al., 2018). Se utilizó el análisis permutacional de dispersiones multivariadas (PERMDISP) para probar la homogeneidad de la dispersión para cada categoría de metadatos (función betadisper del paquete R vegano). Dado que se confirmó la heterogeneidad de la dispersión, se realizó un análisis de similitudes (ANOSIM; función del paquete R vegano).
El método de tamaño del efecto del análisis discriminante lineal (LEfSe) se realizó en datos no normalizados (recuentos brutos de ASV) utilizando la plataforma de analistas de microbiomas (Dhariwal et al., 2017). Los géneros con mayores abundancias relativas en los diferentes sitios de muestreo de la cadena de valor fueron identificados por LEfSe. El efecto del tamaño de cada uno de estos géneros se calculó mediante análisis discriminante lineal (LDA; Segata et al., 2011). Una puntuación LDA (Registro10) de 1,0 se utilizó como punto de corte para la identificación de biomarcadores. LEfSe también se utilizó para comparar las muestras de Fc entre granjas con el fin de identificar bacterias que podrían estar relacionadas con la producción de ácidos grasos de cadena corta. Aquí, se utilizó un LDA de 2 como valor de corte.
El seguimiento de la fuente microbiana se logró para muestras de Dc, Cc y S utilizando el paquete de software SourceTracker (versión 1.0.1) con los parámetros predeterminados (Knights et al., 2011). Se utilizó un valor de rarefacción de 1.000 lecturas y un alfa 1 y 2 de 0,001. Las muestras de granja (Ar, Fc, Sa, Fe) y ambiente (Dev, Cev, Gev, Dcu, Ccu) se consideraron muestras de origen y las de carne (Dc, Cc, S) se consideraron muestras de sumidero.
3 Resultados
3.1 Caracterización de las granjas muestreadas
La microbiota agrícola se caracterizó mediante el análisis de muestras de Ar, Fc, Sa y Fe utilizando amplicones del gen 16S rRNA. A nivel de género, las muestras de Ar fueron similares a Fc, mientras que las muestras de Sa fueron similares a Fe (Figura 1). Al comparar las dos granjas para cada tipo de muestra, la microbiota de Fe, y en menor medida Sa, mostraron diferencias notables. Curiosamente, a nivel de phyla, Proteobacteria representó el 90,7% de las lecturas en Fe de granja-L y 57,6% de granja-H. Las proteobacterias fueron dos veces más abundantes en las muestras de Sa de la granja-H en comparación con la granja-L (Figura suplementaria S2).
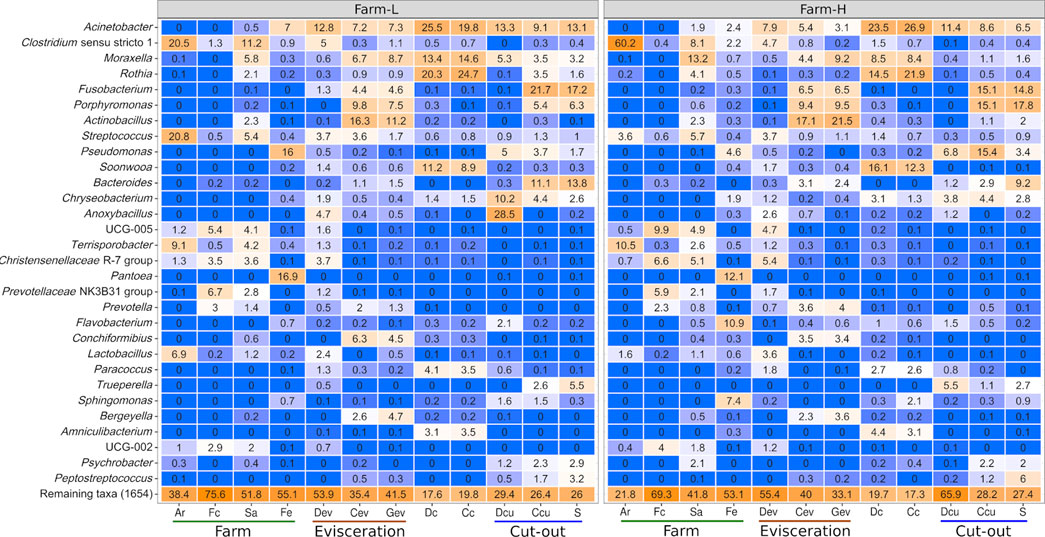 FIGURA 1. Los 30 géneros principales basados en la abundancia relativa (%) identificados en cada uno de los tipos de muestra para ambas granjas. Los géneros se identificaron utilizando la base de datos SILVA. Los gradientes de color varían de azul = 0% a naranja = 100%. Las muestras se recogieron del aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), canales vestidas (Dc), canales frías (Cc), drenaje en el corte (Dcu), transportador en el corte (Ccu) y hombro (S). Granja-L con un estado sanitario más bajo y granja-H con un estatus sanitario más alto, respectivamente.
FIGURA 1. Los 30 géneros principales basados en la abundancia relativa (%) identificados en cada uno de los tipos de muestra para ambas granjas. Los géneros se identificaron utilizando la base de datos SILVA. Los gradientes de color varían de azul = 0% a naranja = 100%. Las muestras se recogieron del aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), canales vestidas (Dc), canales frías (Cc), drenaje en el corte (Dcu), transportador en el corte (Ccu) y hombro (S). Granja-L con un estado sanitario más bajo y granja-H con un estatus sanitario más alto, respectivamente.
Se midieron las concentraciones de ácidos grasos de cadena corta en las heces de animales de ambas granjas y se compararon para evaluar la actividad microbiana (Tabla 1). La concentración de AGCC totales fue significativamente mayor en las muestras de la granja-L que en la granja-H (p = 0,02), así como para cada AGCC individual (todos p < 0,01). Además, los resultados del análisis LEfSe indican una mayor abundancia (p < 0,05, límite de puntuación LDA de 2; datos no mostrados) de géneros específicos conocidos por producir AGCC en granja-L Selenomonas (Wang et al., 2012), Anaerovibrio (Holman et al., 2021), Roseburia (Puertollano et al., 2014; Wang et al., 2019; Markowiak-Kopeć y Śliżewska, 2020), Akkermansia (Li et al., 2021) y Clostridium (Puertollano et al., 2014; Wang et al., 2019; Markowiak-Kopeć y Śliżewska, 2020).
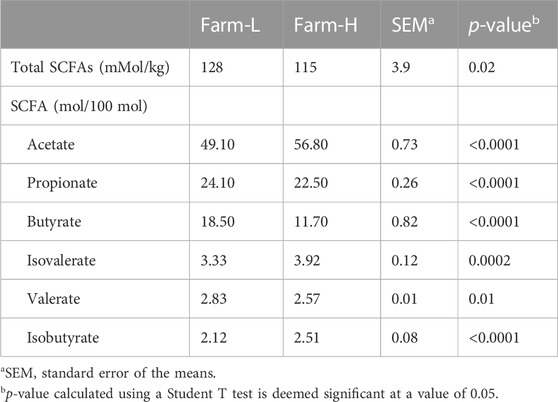 TABLA 1. Concentraciones medias de ácidos grasos de cadena corta (AGCC) en las heces de cerdos de dos granjas con diferentes niveles sanitarios.
TABLA 1. Concentraciones medias de ácidos grasos de cadena corta (AGCC) en las heces de cerdos de dos granjas con diferentes niveles sanitarios.
Se realizó un análisis metabolómico en heces para identificar posibles marcadores metabólicos relacionados con la granja de origen y sus diferentes estados sanitarios. Hubo una clara separación de los datos de las dos granjas en el primer componente principal de la gráfica de puntuación PCA para ionización positiva (27,1%) y negativa (28,5%; Figura suplementaria S3). Esto indica que el entorno y las características de la granja tuvieron una influencia importante en la composición de metabolitos de las heces. El análisis discriminante a través de diagramas de volcanes proporciona otra indicación clara de los diferentes metabolitos que caracterizan cada granja. Bajo ionización positiva, 85 iones fueron significativamente más abundantes en las heces recolectadas de cualquiera de las granjas (Figura 2A). Bajo ionización negativa, 16 iones fueron significativamente más abundantes (Figura 2B). Se obtuvo una identificación putativa basada en masa molecular y espectros de fragmentación para 21 metabolitos en ionización positiva y seis metabolitos en ionización negativa (Tabla Suplementaria S1). Dado que las granjas alimentadas con animales con diferentes formas de dieta (pellet vs. puré) antes del sacrificio y solo se tomaron muestras de dos granjas, no pudimos atribuir con confianza biomarcadores específicos al estado de salud de los animales. No obstante, representan candidatos potenciales para estudios posteriores. En general, nuestros resultados indican que la microbiota de los animales fue diferente entre las dos granjas.
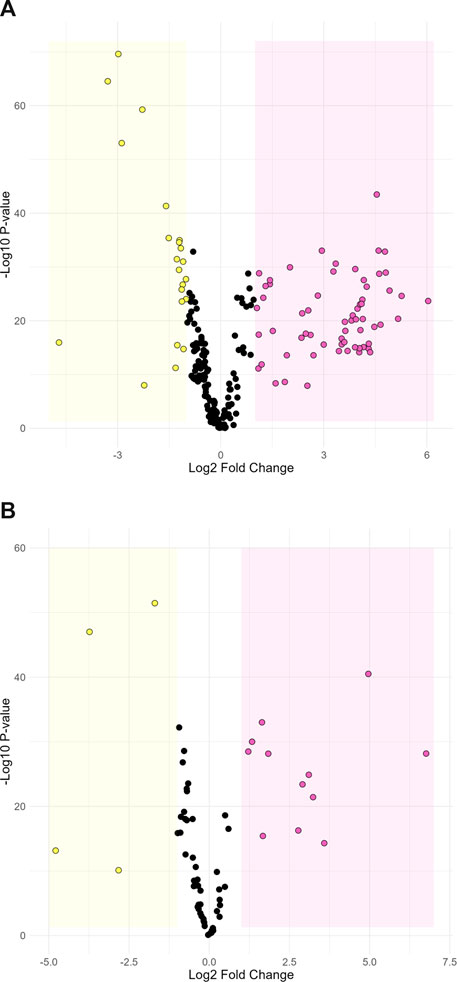 FIGURA 2. Diagrama de volcán que compara la abundancia de metabolitos entre la granja-L y la granja-H bajo ionización positiva (A) y negativa (B). Los puntos en la sección amarilla son iones que son significativamente más abundantes en las muestras de granja-H y los puntos en la sección rosa son significativamente más abundantes en las muestras de granja-L. Las secciones resaltadas representan un registro2-cambio de pliegue mayor que 1 y un valor p menor que 0,05.
FIGURA 2. Diagrama de volcán que compara la abundancia de metabolitos entre la granja-L y la granja-H bajo ionización positiva (A) y negativa (B). Los puntos en la sección amarilla son iones que son significativamente más abundantes en las muestras de granja-H y los puntos en la sección rosa son significativamente más abundantes en las muestras de granja-L. Las secciones resaltadas representan un registro2-cambio de pliegue mayor que 1 y un valor p menor que 0,05.
3.2 Caracterización de la planta cárnica previa a las operaciones
Al comienzo de cada día de sacrificio y despiece de canales, antes de que los animales entren en la línea de procesamiento, la mayoría de las muestras del entorno de la planta cárnica (Cev, Gev, Dcu, Ccu) tenían recuentos microbianos (TAM y EB) alrededor o por debajo del nivel de detección (<2.48 Log10 UFC/300 ml o cm2). Esto fue cierto para todas las semanas de muestreo para ambas granjas. Además, no hubo diferencias significativas (prueba t y Wilcoxon; p > 0.05) en la contaminación inicial de la planta de carne en los días en que se procesaron los animales de cualquiera de las granjas. Para muchas de las muestras, el ADN no pudo extraerse en cantidad suficiente para ser analizado debido a los bajos niveles de contaminación. Un área de muestreo mucho mayor que 300 cm2 habría sido necesario obtener suficiente ADN. A pesar de las similitudes descritas anteriormente, las muestras de Dev tenían medias de recuento TAM de 5,17 ± 1,58 y 2,96 ± 0,24 logaritmos10 CFU/300 mL y EB medias de 3.93 ± 1.35 y 2.88 ± 0.70 Log10 UFC/300 ml para granja-L y granja-H, respectivamente. Los recuentos iniciales para Dev parecen variar sustancialmente entre los días de muestreo después de los procedimientos previos a la operación.
3.3 Contaminación microbiana por presencia de animales en la línea de procesamiento
Como era de esperar, a medida que los animales pasaban por la línea de procesamiento, la UFC en muestras ambientales aumentó significativamente. Para los recuentos de TAM, el aumento fue significativo (p < 0,05) para cada muestra. El EB cuenta en el Gev (p = 0,08) y el drenaje en el corte (Dcu;p = 0,4) las muestras no fueron significativamente diferentes de las recogidas de la línea de procesamiento limpio.
Para evaluar la contaminación de la línea de procesamiento debido a los animales, los recuentos microbianos de las muestras ambientales recolectadas después de que los animales en estudio fueron procesados se restaron de los medidos al final de los procedimientos previos a la operación. No se detectaron diferencias significativas (prueba t y Wilcoxon) entre las granjas para ninguno de los recuentos microbianos (Figura 3). Sólo hubo una tendencia marginal para el recuento de TAM de canales frías (p = 0,067; t-test), donde farm-L tuvo una media de 4,48 ± 0,24 Log10 UFC/300 cm2 y la granja-H tuvo una media de 4,96 ± 0,24 Log10 UFC/300 cm2, pero la diferencia fue inferior a 1 Log10 unidad. Para las muestras de Dc, Cc y S, los recuentos de TAM disminuyeron a medida que las muestras se recolectaron a lo largo de las líneas de procesamiento. Las medias disminuyeron de 4,74 (Dc) y 4,48 (Cc) a 3,08 (S) Log10 UFC/300 cm2 para granja-L, y 4,76 (Dc) y 4,96 (Cc) a 3,39 (S) Log10 UFC/300 cm2 para la granja-H. Sin embargo, los recuentos de EB no siguieron esta tendencia. Las muestras aumentaron de una media de 1,59 (Dc) y 1,33 (Cc) a 2,52 (S) Log10 UFC/300 cm2 para granja-L y 1,71 (Dc) y 1,43 (Cc) a 1,97 (S) Log10 UFC/300 cm2 para la granja-H.
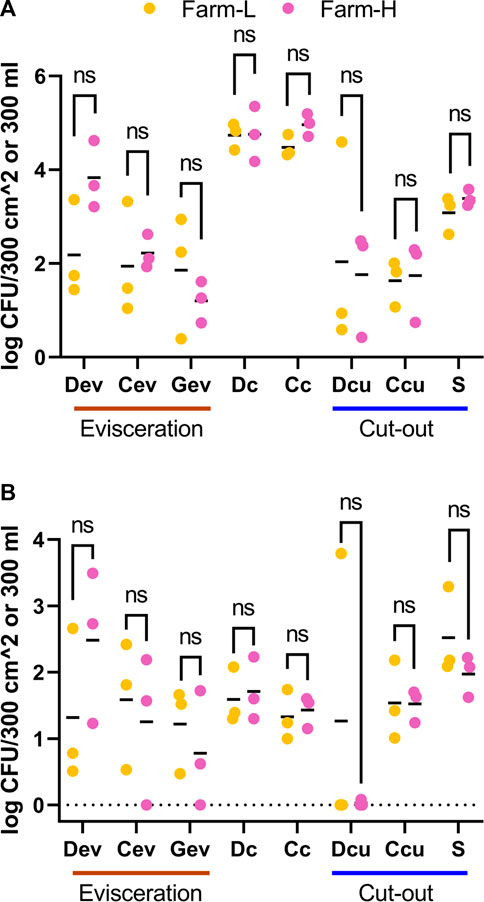 FIGURA 3. Recuentos totales de bacterias mesófilas aerobias (A) y Enterobacteriaceae (B) en Log10 UFC/300 cm2 o 300 ml para muestras asociadas con granja-L (amarillo) o granja-H (rosa), con estatus sanitario más bajo y más alto, respectivamente. Las muestras se recogieron del aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), canales vestidas (Dc), canales frías (Cc), drenaje en el corte (Dcu), transportador en el corte (Ccu) y hombro (S) después de que se procesó el último animal en estudio. Los resultados son la contaminación de los animales, calculada restando los valores obtenidos para la línea de producción limpia. El nivel de detección para muestras de drenaje fue de ≤2,48 Log10 UFC/300 ml para TAM y EB. Para las otras muestras, el nivel de detección fue ≤1.22 Log10 UFC/300 cm2 para TAM y ≤0.70 Log10 UFC/300 cm2 para EB.
FIGURA 3. Recuentos totales de bacterias mesófilas aerobias (A) y Enterobacteriaceae (B) en Log10 UFC/300 cm2 o 300 ml para muestras asociadas con granja-L (amarillo) o granja-H (rosa), con estatus sanitario más bajo y más alto, respectivamente. Las muestras se recogieron del aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), canales vestidas (Dc), canales frías (Cc), drenaje en el corte (Dcu), transportador en el corte (Ccu) y hombro (S) después de que se procesó el último animal en estudio. Los resultados son la contaminación de los animales, calculada restando los valores obtenidos para la línea de producción limpia. El nivel de detección para muestras de drenaje fue de ≤2,48 Log10 UFC/300 ml para TAM y EB. Para las otras muestras, el nivel de detección fue ≤1.22 Log10 UFC/300 cm2 para TAM y ≤0.70 Log10 UFC/300 cm2 para EB.
Los inspectores recopilaron varios datos no microbiológicos para evaluar el peso de la canal y otros defectos potenciales (véase la descripción anterior; no se muestran los datos). No se observaron diferencias significativas entre las dos granjas para ninguno de los datos de inspección recopilados.
3.4 Análisis de las variaciones de la microbiota en toda la cadena de valor porcina utilizando amplicones de ADNr 16S
En general, Acinetobacter fue el género más abundante en todas las muestras, excepto en Ar y Fc (Figura 1). Estas bacterias se encontraron principalmente en la superficie de canales vestidas (Dc) y frías (Cc; entre 20% y 27% de abundancia relativa) y se detectaron en menor abundancia en todas las muestras ambientales de plantas cárnicas. Clostridium (Clostiridum_sensu_stricto_1) fue particularmente abundante en muestras de Ar (20,5% y 60,2% para granjas-L y H, respectivamente). Fusobacterium, Porphyromonas y Bacteroides se detectaron principalmente en superficies de equipos (Cev, Gev y Ccu) y muestras S. Los resultados variaron para las muestras de Ccu y S dependiendo de la granja. Streptococcus fue el único género encontrado en toda la cadena de valor. Este género generalmente tenía bajas abundancias (<5,7%) excepto para las muestras de aire de granja L (20,8%). La distribución de Pseudomonas varió mucho entre las muestras. En las muestras de Fe, Pseudomonas fue más abundante en la granja-L (16,0%) que en la granja-H (4,6%). Lo contrario se encontró para Ccu (3,7% en la granja-L, 15,4% en la granja-H). La abundancia de Anoxybacillus fue diferente entre las dos granjas para Dcu (28.5% en granja-L y 1.2% en granja-H). La alta abundancia relativa media de Anoxybacillus en la granja-L fue causada por una muestra con abundancia extremadamente alta. Este resultado fue consistente con los recuentos anormalmente altos de placas para TAM y EB en la misma muestra contaminada (Figura 3; Dcu).
Ciertas familias bacterianas tienen un gran impacto en la cadena de valor de la carne de cerdo desde una perspectiva de seguridad e higiene de la carne. Algunos causan deterioro de la carne, mientras que otros aumentan la vida útil de la carne. Cinco de esas familias, Campylobacteraceae, Carnobacteriaceae, Enterobacteriaceae, Lactobacillaceae y Staphylococcaceae, se examinaron como un subconjunto de los datos totales. Las abundancias relativas de los géneros dentro de esta submuestra se presentan en la Figura 4. No se detectaron Listeriaceae. Las abundancias de estas familias eran similares entre las dos granjas. Campylobacteraceae se encontraron principalmente en muestras de Fc, Cev, Gev y S. Las carnobacterias se detectaron principalmente en el área de corte, pero también se encontraron en toda la cadena de valor. Sólo se encontraron rastros en Fc y Fe. Las enterobacteriaceae estaban presentes solo en pequeñas cantidades en muestras de AR. Se identificaron altas abundancias de Lactobacillaceae en Ar, Fc, Sa, Dev y en superficies de canales. Staphylococcaceae se encontraron en toda la cadena de valor, pero solo en pequeñas cantidades en Fc y Dev. A nivel de género, Allicoccus, Atlantibacter, Companilactobacillus, Corticicoccus, Cronobacter, Mamallicoccus, Serratia, Salinicoccus, Salmonella y Weisella (p < 0.001) fueron significativamente más abundantes en muestras de granja-L. El único género que fue más abundante en las muestras de granja-H fue Lacticigenium (p < 0,001; Figura suplementaria S4). Se detectaron Escherichia/Shigella en ambas explotaciones principalmente en Sa y Fc, y en Dev, Cev y Gev. El mismo ASV de Escherichia/Shigella detectado en Sa y Fc también se encontró en el ambiente de la planta cárnica (ASV168; Cuadro suplementario S2). Contrariamente a Campylobacter, solo se detectaron rastros de Escherichia/Shigella en las canales y los cortes de carne de paleta resultantes.
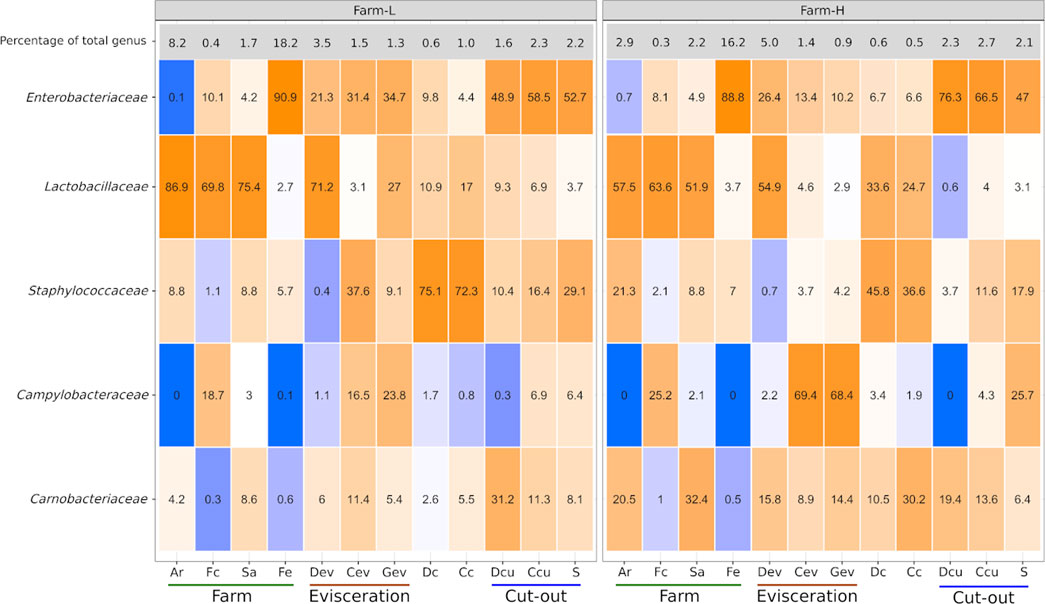 FIGURA 4. Campylobacteraceae, Carnobacteriaceae, Enterobacteriaceae, Lactobacillaceae y Staphylococcaceae abundancias relativas (%) entre sí calculadas para cada uno de los tipos de muestra para ambas granjas. La fila superior indica el porcentaje que estas cinco familias representaron en la microbiota total de cada tipo de muestra. Los gradientes de color varían de azul = 0% a naranja = 100%. Las muestras se recogieron del aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), canales vestidas (Dc), canales frías (Cc), drenaje en el corte (Dcu), transportador en el corte (Ccu) y hombro (S). Granja-L con un estado sanitario más bajo y granja-H con un estatus sanitario más alto, respectivamente.
FIGURA 4. Campylobacteraceae, Carnobacteriaceae, Enterobacteriaceae, Lactobacillaceae y Staphylococcaceae abundancias relativas (%) entre sí calculadas para cada uno de los tipos de muestra para ambas granjas. La fila superior indica el porcentaje que estas cinco familias representaron en la microbiota total de cada tipo de muestra. Los gradientes de color varían de azul = 0% a naranja = 100%. Las muestras se recogieron del aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), canales vestidas (Dc), canales frías (Cc), drenaje en el corte (Dcu), transportador en el corte (Ccu) y hombro (S). Granja-L con un estado sanitario más bajo y granja-H con un estatus sanitario más alto, respectivamente.
Se determinó la diversidad alfa para cada tipo de muestra para evaluar la diversidad microbiana en términos de riqueza (Observado y Chao1) y riqueza y uniformidad (Shannon y Simpson; Figura 5). Hubo diferencias significativas entre las granjas para las muestras de Ar y Fe (p < 0,05). La diversidad fue menor en las muestras de Fe y mayor en las muestras de Sa (p < 0,05; Observado y Chao1). En términos de uniformidad, la microbiota en el Ar fue más uniforme para la granja-L que para la granja-H, mientras que lo contrario se observó para las muestras de Fe. El índice de Simpson para las muestras de Ar fue distinto de los otros tipos de muestra e indicó una clara prevalencia de unos pocos taxones, a saber, Clostridium en granja-H y Clostridium y Streptococcus en granja-L (Figura 1). A medida que se obtuvieron muestras a lo largo de las diferentes ubicaciones y pasos de la cadena de valor, hubo una clara reducción de la diversidad microbiana. Sin embargo, hubo una gran variación en los índices de Shannon y Simpson para la granja-L, lo que sugiere que la microbiota varió sustancialmente entre las tres semanas de envío.
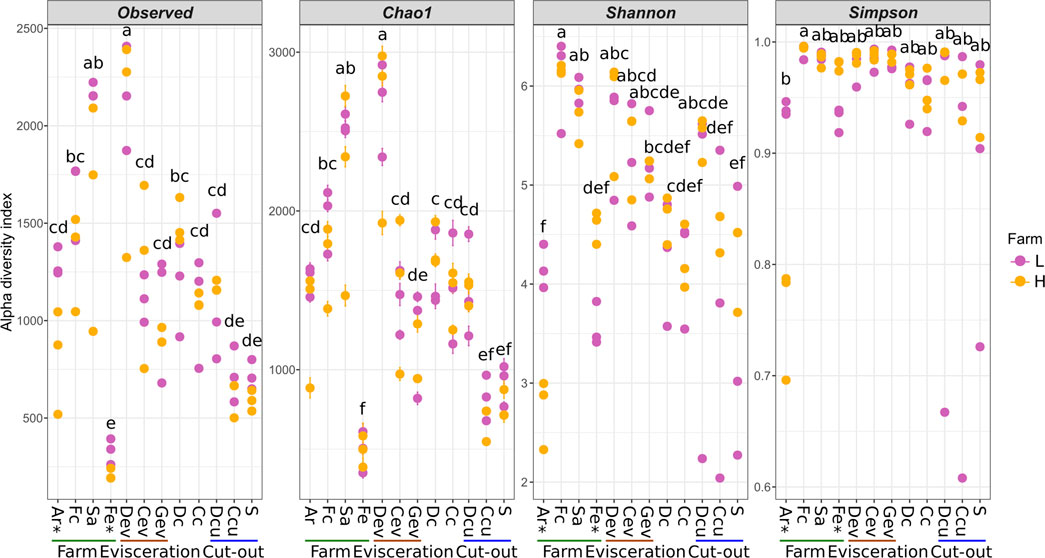 FIGURA 5. Diversidad alfa de las muestras a lo largo de la cadena de valor calculada con diferentes índices de riqueza (Observado, Chao1) y riqueza y uniformidad (Shannon, Simpson). Las diferencias significativas entre las dos granjas según una prueba T de Student (p < 0,05) se identifican con un asterisco (*) en la parte inferior del gráfico. Se identifican muestras significativamente diferentes con diferentes letras de acuerdo con una prueba de Tukey HSD (p < 0.05). Las muestras se recogieron del aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), canales vestidas (Dc), canales frías (Cc), drenaje en el corte (Dcu), transportador en el corte (Ccu) y hombro (S). Granja-L (amarillo) o granja-H (rosa) con estatus sanitario más bajo y más alto, respectivamente.
FIGURA 5. Diversidad alfa de las muestras a lo largo de la cadena de valor calculada con diferentes índices de riqueza (Observado, Chao1) y riqueza y uniformidad (Shannon, Simpson). Las diferencias significativas entre las dos granjas según una prueba T de Student (p < 0,05) se identifican con un asterisco (*) en la parte inferior del gráfico. Se identifican muestras significativamente diferentes con diferentes letras de acuerdo con una prueba de Tukey HSD (p < 0.05). Las muestras se recogieron del aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), canales vestidas (Dc), canales frías (Cc), drenaje en el corte (Dcu), transportador en el corte (Ccu) y hombro (S). Granja-L (amarillo) o granja-H (rosa) con estatus sanitario más bajo y más alto, respectivamente.
Se realizó un análisis de diversidad beta para visualizar las diferencias de diversidad entre muestras e identificar qué factores afectan los cambios en la microbiota a lo largo de la cadena de valor (Tabla 2). Las distancias UniFrac no ponderadas y ponderadas y las diferencias de Bray-Curtis se visualizaron utilizando PCoA (Figura 6) y una prueba ANOSIM (p < 0,05) en las matrices de distancia. En todas las medidas de distancia, el tipo de muestra y la ubicación (granjas, área de evisceración y muestras de área de recorte) fueron significativos (p < 0.0001), mientras que las granjas (L y H) y las semanas de envío (1, 2 y 3) no lo fueron. El tipo de muestra tuvo el valor R más alto, lo que indica que explicó la mayor cantidad de variación. Cada tipo de muestra tenía una microbiota única. Luego se reagruparon en función de su posición a lo largo de la cadena de valor (Figura 6). Esta agrupación secundaria está definida por la microbiota única en cada granja, el área de evisceración y el área de corte. En todas las métricas de distancia, las muestras se distribuyeron principalmente a lo largo de la coordenada principal 1 (PCo1) según su ubicación, lo que indica que la microbiota en las granjas se reemplaza y se vuelve menos diversa más abajo en la cadena de valor. Las únicas excepciones fueron las muestras Dev y Fe. Las muestras de Dev variaron entre granjas y estaban distantes de las otras muestras de la planta de carne. Las muestras de Fe fueron más similares a las muestras de la planta de carne que a otras muestras de las granjas. Las coordenadas principales 2 (PCo2) y 3 (PCo3) parecen estar relacionadas principalmente con las diferencias entre los diferentes tipos de muestra. Cuando se considera solo la abundancia (diferencias de Bray-Curtis; Figuras 6A, B), la microbiota PCo2 estuvo influenciada principalmente por las diferencias entre Cev y Gev, que es máxima en comparación con Dcu. Para PCo3, las diferencias entre las muestras de canales (Dc y Cc) y el resto de las muestras de plantas de carne fueron evidentes. Cuando solo se considera la filogenia (distancias UniFrac no ponderadas; Figuras 6C, D) a lo largo del PCo2, las muestras se distribuyeron principalmente en función de la disimilitud entre el Fe y las muestras de plantas cárnicas. En el eje PCo3, las diferencias entre el DCU y el resto de las muestras fueron evidentes. Cuando se considera tanto la abundancia como la filogenia (distancias UniFrac ponderadas; Figuras 6E, F), las muestras se distribuyeron principalmente a lo largo del PCo2 según sus similitudes con las muestras Ar o Cev y Gev. A lo largo del PCo3, se ilustran variaciones más pequeñas (el eje es más estrecho) a través de pequeñas diferencias entre las muestras de Fe y Ar.
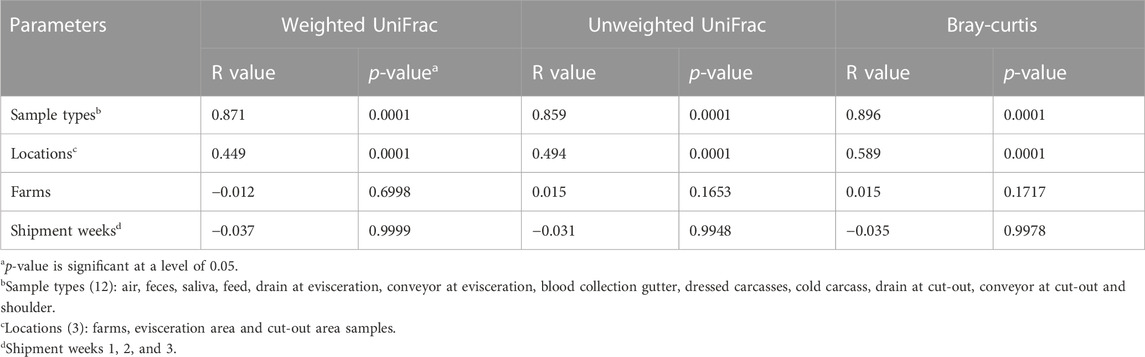 TABLA 2. Factores asociados con la estructura de la comunidad de la microbiota para muestras recogidas a lo largo de la cadena de valor porcina medida utilizando ANOSIM de las distancias UniFrac ponderadas y no ponderadas y las diferencias de Bray-Curtis (valor R).
TABLA 2. Factores asociados con la estructura de la comunidad de la microbiota para muestras recogidas a lo largo de la cadena de valor porcina medida utilizando ANOSIM de las distancias UniFrac ponderadas y no ponderadas y las diferencias de Bray-Curtis (valor R).
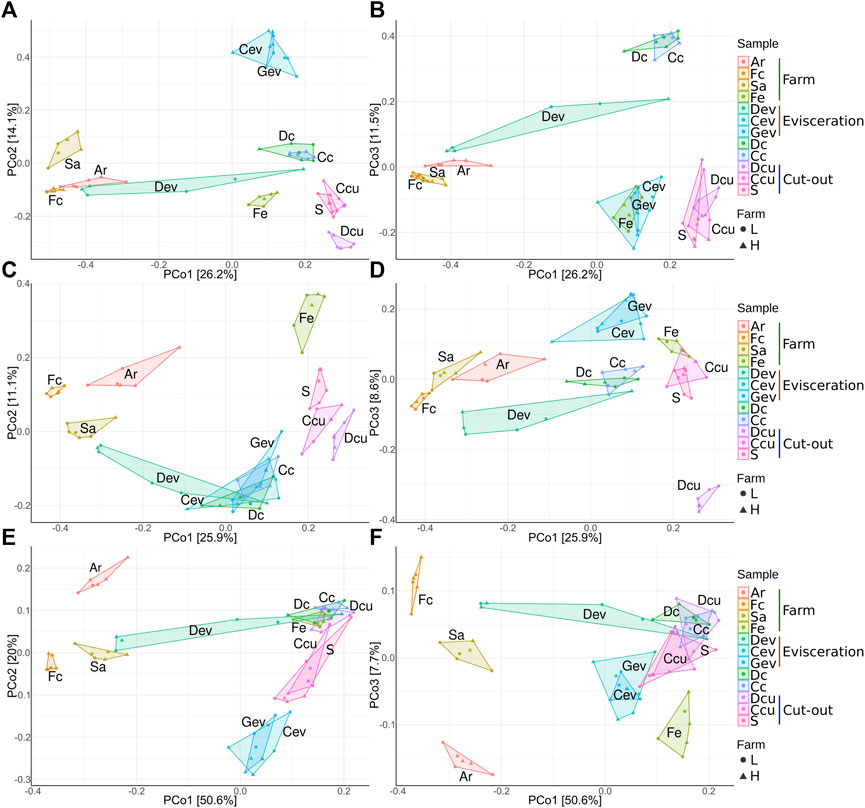 FIGURA 6. Gráficos de análisis de coordenadas principales de disimilitudes de Bray-Curtis (A, B), distancias UniFrac no ponderadas (C, D) y distancias UniFrac ponderadas (E, F) clasificadas por tipo de muestra. El lado derecho (A, C, E) son los ejes 1 y 2 y el lado izquierdo (B, D, F) son los ejes 1 y 3. Las muestras se recogieron del aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), canales vestidas (Dc), canales frías (Cc), drenaje en el corte (Dcu), transportador en el corte (Ccu) y hombro (S). Granja-L o granja-H con estatus sanitario más bajo y más alto, respectivamente.
FIGURA 6. Gráficos de análisis de coordenadas principales de disimilitudes de Bray-Curtis (A, B), distancias UniFrac no ponderadas (C, D) y distancias UniFrac ponderadas (E, F) clasificadas por tipo de muestra. El lado derecho (A, C, E) son los ejes 1 y 2 y el lado izquierdo (B, D, F) son los ejes 1 y 3. Las muestras se recogieron del aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), canales vestidas (Dc), canales frías (Cc), drenaje en el corte (Dcu), transportador en el corte (Ccu) y hombro (S). Granja-L o granja-H con estatus sanitario más bajo y más alto, respectivamente.
El análisis LEfSe se realizó en un esfuerzo por identificar géneros que podrían usarse como biomarcadores de la granja de origen. Ningún género se asoció fuertemente con ninguna de las granjas utilizando parámetros LEfSe estándar (p < 0,05 para la prueba de Kruskal-Wallis y el corte de puntuación LDA de 2). Cuando la gravedad del límite de la puntuación LDA se redujo a 1, 12 géneros se asociaron con una granja específica (Figura 7). Flavobacterium y Eubacterium_saphenum_group se asociaron con la granja H. Anaevibrio, Mageeibacillus, Micrococcus, Megasphaera, Akkermansia, Selenomonas, Ewingella, Deinococcus, Bifidobacterium y Mistsuokella se asociaron con farm-L. Los géneros asociados con la granja-L se encontraron en toda la cadena de valor, mientras que los biomarcadores para la granja-H estaban presentes principalmente en las muestras de plantas cárnicas. Estos géneros representaron un pequeño porcentaje (<4,5%) de la abundancia relativa total en todos los tipos de muestras. La única excepción son las muestras de Fe, donde constituyen un porcentaje mayor (8,3% en la granja-L y 10,9% en la granja-H; Figura complementaria S5). De los géneros identificados, sólo Anaevibrio, Mageeibacillus, Micrococcus, Megasphaera, Akkermansia y Flavobacterium estaban por encima del umbral de 1,5 LDA puntuación. Ninguno de los géneros identificados aquí tuvo una puntuación LDA de 2 o superior. Esto indica que mientras que la prueba de Kruskal-Wallis enumera estas bacterias como significativamente más abundantes en una de las granjas, el análisis discriminante lineal muestra que tenían baja relevancia como biomarcadores del origen de la granja. Inversamente, esto significa que las bacterias que impactan la cadena de valor no se vieron afectadas por la granja con la que estaban asociadas. Este hallazgo es consistente con los resultados del análisis ANOSIM (Tabla 2). Sin embargo, dado que solo dos granjas se procesaron en una planta, eso no significa que sea posible, para todas las organizaciones comerciales, producir productos de calidad microbiana de carne similares a partir de animales procedentes de dos granjas diferentes.
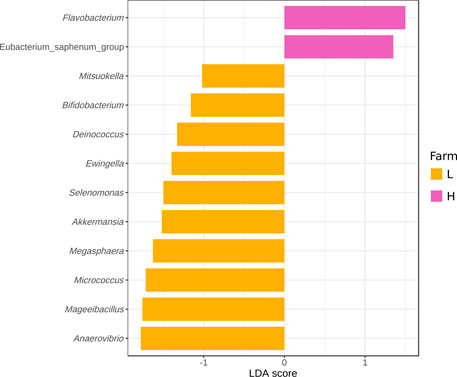 FIGURA 7. Géneros diferencialmente abundantes en todas las muestras evaluados mediante análisis discriminante lineal (LDA) con mediciones de tamaño de efecto (LEfSe) para granja-L (amarillo) y granja-H (rosa) con estados sanitarios más bajos y más altos, respectivamente. Sólo aquellos géneros con una puntuación LDA (log10) de >1.0.
FIGURA 7. Géneros diferencialmente abundantes en todas las muestras evaluados mediante análisis discriminante lineal (LDA) con mediciones de tamaño de efecto (LEfSe) para granja-L (amarillo) y granja-H (rosa) con estados sanitarios más bajos y más altos, respectivamente. Sólo aquellos géneros con una puntuación LDA (log10) de >1.0.
Se utilizó un análisis de seguimiento de la fuente para identificar posibles fuentes de contaminación microbiana en las canales y los cortes de hombros. Como se muestra en la Figura 8A, la mayoría de los microorganismos encontrados (como ASV) en muestras de Dc probablemente no se originaron en la granja ya que se les atribuyeron ASV limitados. Las muestras de granja (Ar, Fc, Sa y Fe) son la fuente de solo el 2.2% de los ASV que también se encuentran en las muestras de Dc. Las fuentes de contaminación siguen siendo en su mayoría desconocidas (81%), pero el 14,4% provino de las muestras de Dev. La Figura 8B muestra que la refrigeración nocturna después del enfriamiento inicial no cambió la microbiota de las canales de manera importante, ya que el 89,9% de los ASV fueron los mismos en las muestras de Cc y CC. La figura 8C indica que la mayoría de los microorganismos detectados en las muestras de S probablemente se originaron a partir de Cc (79,6%). Las muestras de granja (Ar, Fc, Sa y Fe) representaron un total de 3.52% de ASV compartidos y la mayoría de las muestras de plantas de carne (Dev, Cev, Gev, Dcu y Ccu) representan 9.39%.
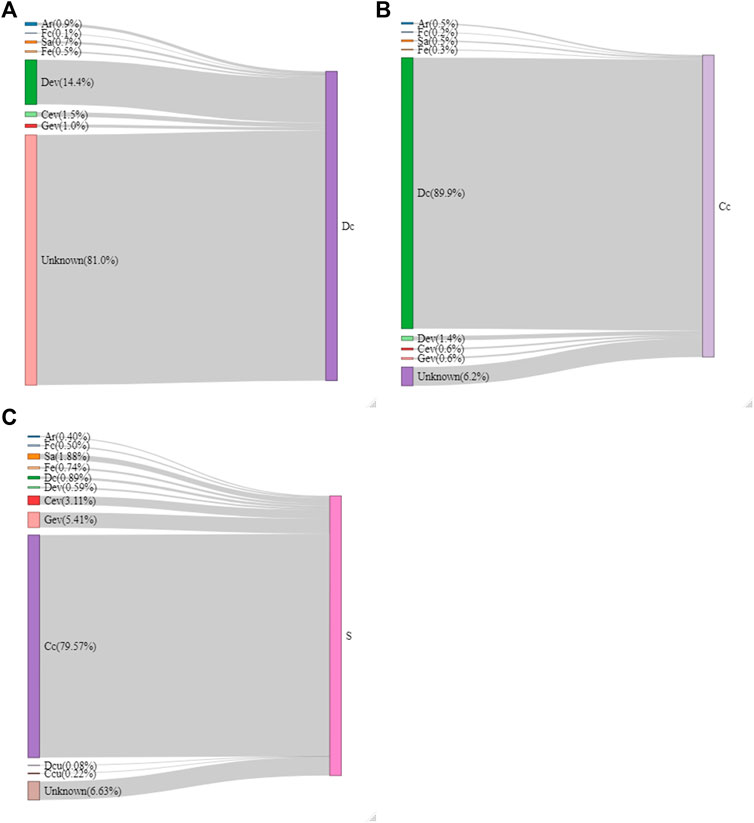 FIGURA 8. Diagrama de flujo de las fuentes de microorganismos en la canal vestida [Dc; A)], canal fría [Cc; (B)] y el hombro [S; (C)] muestras generadas por el software SourceTracker. La proporción (%) que representa cada fuente en la muestra de carne seleccionada se indica entre paréntesis. Las muestras se recogieron de aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), drenaje en el corte (Dcu), transportador en el corte (Ccu).
FIGURA 8. Diagrama de flujo de las fuentes de microorganismos en la canal vestida [Dc; A)], canal fría [Cc; (B)] y el hombro [S; (C)] muestras generadas por el software SourceTracker. La proporción (%) que representa cada fuente en la muestra de carne seleccionada se indica entre paréntesis. Las muestras se recogieron de aire (Ar), heces (Fc), saliva (Sa), alimento (Fe), drenaje en la evisceración (Dev), transportador en la evisceración (Cev), canalón de recolección de sangre (Gev), drenaje en el corte (Dcu), transportador en el corte (Ccu).
4 Discusión
En este estudio, se compararon las diferencias en la microbiota en muestras tomadas de múltiples pasos a lo largo de la cadena de valor (desde la granja hasta el área de corte de carne) para dos granjas con diferentes estados sanitarios. Uno de los objetivos era medir la contribución de la granja y la planta cárnica a la microbiota que se encuentra en los cortes de carne. La fuerza de nuestro diseño experimental proviene del hecho de que los mismos animales que fueron criados en granjas comerciales fueron seguidos a lo largo de toda la cadena de valor.
Sobre la base de los resultados metataxonómicos, la concentración de AGCC y el análisis metabolómico de las heces, las dos granjas se distinguieron en términos de su microbiota. Esto es consistente con las observaciones hechas por los veterinarios que seleccionaron las granjas. Se ha demostrado que las condiciones de higiene en las granjas afectan a la microbiota en el intestino de los cerdos (Le Floc’h et al., 2014), su alimentación (Maciorowski et al., 2007) y el aire (Duchaine et al., 2000). Los resultados indican que los géneros bacterianos identificados en las muestras de Ar y Fc estaban estrechamente relacionados como se esperaba, ya que las partículas fecales secas son una fuente importante de contaminación del aire (Hamscher et al., 2003). Los problemas mecánicos con el sistema de ventilación en la granja-H podrían explicar parte de la variación observada en la microbiota del aire. Se sabe que la ventilación ineficiente admite concentraciones más altas de microbios totales y porcentajes más altos de microbios fecales en el aire (Kim y Ko, 2019). Estos microbios, de los cuales Clostridium es una gran parte (Nehme et al., 2008; Song et al., 2021), distinguieron claramente las dos granjas (Figura 1). La alta diversidad alfa en el aire en los edificios donde se crían cerdos se ha relacionado con una mayor incidencia de genes de resistencia a los antibióticos y patógenos oportunistas (Kumari et al., 2016; Yan et al., 2021). En nuestro estudio, los recuentos de ASV en muestras de Ar fueron más altos que los reportados en la literatura (Kraemer et al., 2019; Yan et al., 2021). La alta diversidad alfa observada/ChaO1 que observamos puede deberse, en parte, a la agrupación de todas las muestras en la etapa de inferencia de muestras de la tubería bioinformática, lo que puede aumentar la sensibilidad a los ASV raros.
Debido a que el alimento entra en contacto con la saliva, se espera que su composición bacteriana se superponga y cambie con el tipo de alimento. Un mes antes del muestreo, farm-L experimentó un brote de salmonelosis, lo que requirió un cambio a alimento para puré para controlar la diarrea causada por el brote (Lo Fo Wong et al., 2004; Mikkelsen Lene y otros, 2004; Longpré y otros, 2016; Lebel et al., 2017; O’Meara et al., 2020). Se sabe que el alimento con puré induce niveles más altos de AGCC en las heces, lo que a su vez mejora el perfil hematológico de los cerdos y reduce la ulceración, los niveles de diarrea, así como el desprendimiento de Salmonella y E. coli (Lo Fo Wong et al., 2004; Mikkelsen Lene y otros, 2004; Longpré y otros, 2016; Lebel et al., 2017; O’Meara et al., 2020). Nuestro análisis de Lefse confirmó que el alimento para puré promovió bacterias productoras de AGCC bien conocidas, probablemente causando la mayor concentración de AGCC en las heces de animales de granja-L. Esto también puede haber contribuido a estabilizar la microbiota, lo que resulta en una diversidad alfa similar a la de las heces de la granja-H. También se sabe que el alimento para puré alberga recuentos bacterianos más altos que el alimento granulado en caliente tradicional (Mikkelsen Lene et al., 2004; Paramithiotis y otros, 2009; O’Meara et al., 2020). Por lo tanto, el tipo de alimento fue un factor importante para determinar las diferencias entre las dos granjas en nuestro estudio.
La planta de carne estaba limpia antes de que cualquiera de los animales en estudio fuera procesado, lo que nos permitió evaluar la contaminación que provenía de los animales. Se ha informado que las muestras ambientales de varios tipos varían de 2 a 6 Log10 de UFC/cm2 para TAM y de no detectado a 5 Log10 de UFC/cm2 para EB (Warriner et al., 2002; Bridier et al., 2019; Maes et al., 2019). Nuestros recuentos microbianos para TAM y EB estaban por debajo de esos valores en muestras de Cev, Gev, Ccu y Dcu. Solo las muestras de Dev mostraron niveles muy variables de contaminación a lo largo del tiempo. Esto es consistente con la literatura que sugiere que los drenajes de las plantas de procesamiento son una fuente de contaminación, especialmente a través de la formación de aerosoles (Byrne et al., 2008), y son un contribuyente principal a la variación de la contaminación y la evolución de una planta de procesamiento a lo largo del tiempo (ICMSF, 2018).
En general, el nivel de contaminación de la planta de carne fue similar después de que los animales en estudio fueron procesados para ambas granjas. Sin embargo, la temporada es un factor importante que contribuye a la contaminación en los entornos de cría de cerdos (Kumari et al., 2016) y su efecto debe abordarse durante un período de tiempo más largo. canal (dc y cc; Figura 2) y muestras de drenaje (Dev y Dcu; Figura 3) tuvieron números similares de TAM y EB en comparación con el rango reportado en la literatura (Spescha et al., 2006; Gill y Badoni, 2010; Zwirzitz et al., 2020), mientras que las muestras de Cev, Gev y Ccu (Figura 3) fueron menores (Warriner et al., 2002; Bridier et al., 2019; Zwirzitz et al., 2020). Los recuentos totales de mesófilos, enterobacteriaceae y coliformes aeróbicos aumentaron en el equipo de procesamiento con el tiempo (Warriner et al., 2002). Los bajos recuentos observados pueden explicarse por el hecho de que los animales examinados fueron los primeros del día en ser sacrificados. Pasaron menos de una hora en la línea de procesamiento, que procesaba 500 animales por hora. Las muestras de Gev y Dcu tuvieron un aumento significativo en TAM después de que los animales fueron procesados (Figura 3). Este no fue el caso de los recuentos de EB. Gev recolectaba sangre principalmente y si los cerdos estaban sanos, su sangre contenía un número limitado de microbios (Hyun et al., 2021). Esto puede explicar por qué los recuentos de EB no cambiaron durante el corto período de muestreo en muestras de Gev. El agua caliente fluyó a través de DCU durante la operación. Debido a que las enterobacterias no forman esporas (Brenner y Farmer, 2015), es probable que no puedan sobrevivir en esas condiciones. Los recuentos de células sugieren que los riesgos de contaminación son similares para ambas granjas. La abundancia relativa ligeramente mayor de Salmonella en muestras de granja-L podría sugerir lo contrario, aunque solo se detectaron trazas (0.036% de abundancia relativa en Fe y 0.0045% en Cev).
La diversidad alfa disminuyó en las muestras a lo largo de la cadena de valor, lo que sugiere que la microbiota se vuelve menos compleja, especialmente para las muestras Dc, Cc y S. Esta reducción progresiva de la diversidad se explica en parte por el tratamiento térmico (escaldado y chamuscado) que forma parte del procesamiento del animal, y los posteriores procedimientos de depilado y pulido. Estos pasos eliminan una gran parte de la microbiota de la piel que podría ser recontaminada por la propagación de bacterias residentes de la planta de carne a los cadáveres (Gill y Bryant, 1993; Gill, 2000; Wheatley et al., 2014; Zwirzitz et al., 2020). En Zwirzitz et al. (2020), la diversidad alfa disminuyó entre la llegada de los animales al matadero y la chamuscación, aumentó nuevamente en la etapa de pulido y luego disminuyó continuamente hasta que las canales se enviaron a una instalación de corte de carne. Esto indica que el pulido es un paso crítico para reemplazar la microbiota animal en la superficie de las canales con bacterias residentes del matadero. Claramente, los pasos de procesamiento previos a la refrigeración nocturna justifican una mayor investigación. En nuestro estudio, este proceso podría explicar cómo la diversidad beta de los cadáveres de ambas granjas no fue significativamente diferente. El tipo de muestra y la ubicación (granja vs. área de evisceración vs. área de corte) afectaron significativamente la estructura de la comunidad de microbiota (Tabla 2).
Hay indicios de que algunas de las bacterias de las granjas todavía estaban presentes en los cadáveres y las muestras de hombro. Escherichia/Shigella se detectaron principalmente en muestras de Sa y Fe en las granjas y en muestras de Dev, Cev y Gev en el área de evisceración. El mismo ASV de Escherichia/Shigella detectado en muestras de Sa y Fc también se encontró en todas las muestras de plantas cárnicas (ASV168; Cuadro suplementario S2) y se detectaron trazas en muestras Cc y S. Campylobacter, el patógeno más común en la cadena de valor porcina (Farzan et al., 2010; Baer et al., 2013), se detectó en muestras Sa, Fe y S. Los datos revelaron que los ASV detectados en las muestras S no eran los mismos que los encontrados en Sa, Fe, Sa, Dev y Dcu (Tabla Suplementaria S2) y, en cambio, eran los mismos que Cev, Gev y Ccu. Esto sugiere que una parte del Campylobacter detectado podría ser de superficies de plantas cárnicas. Además, Carnobacterium se detectó casi exclusivamente en el lugar de corte, lo que indica que estas bacterias son de la planta de carne. Estas bacterias están casi ausentes en los cadáveres, pero se identificaron en muestras S. Curiosamente, este género, junto con muchas Enterobacteriaceae (Figura Suplementaria S2; Enterobacter, Citrobacter, Buttilauxella, Lelliottia y Kluyvera), reemplazaron a Lactobacillus, Moraxella y Staphylococcus en muestras de S. Esto podría explicar por qué los recuentos de enterobacteriaceae aumentaron en los hombros en lugar de seguir la tendencia general a la baja de los recuentos mesófilos aeróbicos totales después de la descomposición de la canal. Múltiples estudios (Gill y Bryant, 1993; Gill y otros, 1999; Gill, 2000; Gill y Sofos, 2005) investigó el impacto de la descomposición de las canales en la microbiota de la carne. Se transfieren múltiples tipos de bacterias desde el equipo, los trabajadores, etc. a los cortes de carne resultantes durante los pasos de procesamiento en la cadena de valor, reemplazando aún más cualquier bacteria de la granja. El análisis de seguimiento de la fuente confirma que solo el 12,92% de la microbiota en muestras S se originó en las granjas y el entorno de la planta cárnica (Figura 8C).
En general, el 81% de las bacterias detectadas en las muestras de Dc eran de origen desconocido (Figura 8A), lo que indica que queda mucho por investigar para comprender las fuentes de contaminación encontradas en los cortes finales de carne. Los procesos de depilación y pulido serían pasos valiosos a considerar para determinar las fuentes de contaminación (Gill, 2000). Una vez que los cadáveres se enfriaron rápidamente, la microbiota fue menos susceptible a la variación entre las muestras de Dc, Cc y S; El 89,9% de las bacterias encontradas en las muestras de Cc provenían del Dc y el 79,6% de las bacterias encontradas en las muestras de S provenían de Cc. La refrigeración favorece el desarrollo de una microbiota más psicrotrófica (Zwirzitz et al., 2020) y en las muestras S se detectaron algunas bacterias tolerantes al frío, ya que el área de corte se mantuvo.
5 Conclusión
La variabilidad en la microbiota observada en las muestras a lo largo de la cadena de valor estuvo más asociada con la ubicación (granja, área de evisceración, área de recorte) y el tipo de muestra que con la granja de origen o la semana de envío. Los microbios identificados en cada granja fueron controlados en gran medida por procedimientos operativos internos. Esto se evidencia por el hecho de que menos del 4% de las bacterias identificadas en las muestras S, el último paso en la cadena de valor que se examinó, se determinó que eran de las granjas. De hecho, se observó una microbiota más homogénea y menos compleja a medida que avanzamos en la cadena de valor. Sin embargo, sería presuntuoso afirmar que las plantas cárnicas descontaminan completamente a los animales de su microbiota inicial adquirida en la granja, independientemente de su estado sanitario, ya que se encontró Salmonella en baja abundancia relativa con muestras de Fe y Cev asociadas a la granja-L. Los resultados sugieren más bien que el control microbiano que actualmente logran las granjas permite producir productos porcinos con una calidad microbiológica adecuada. Sin embargo, se necesita un estudio más amplio que incluya más granjas con diferentes estados sanitarios para determinar con precisión el impacto y la magnitud del estado sanitario de la granja en los productos porcinos comerciales finales, sabiendo que muchos factores como la temporada, la forma y composición del alimento, las condiciones de alojamiento, el manejo previo al sacrificio, etc., también son factores contribuyentes.
Declaración de disponibilidad de datos
Los conjuntos de datos presentados en este estudio se pueden encontrar en repositorios en línea. Los nombres del repositorio / repositorios y el (los) número (s) de acceso se pueden encontrar en el artículo / Material complementario.
Declaración ética
El estudio en animales fue revisado y aprobado por el Comité de Uso y Cuidado de Animales de la Universidad Laval (2019-329). Se obtuvo el consentimiento informado por escrito de los propietarios para la participación de sus animales en este estudio.
Contribuciones del autor
Conceptualización, LS y SF; metodología, PL, CD, ÉP y PF; software, AV, AD-F, P-LP y PL; validación, LS, P-LP y AV; investigación, PL; recursos, LS y CD; curación de datos, PL, AD-F, P-LP y AV; redacción\u2012preparación original del borrador, PL; redacción\u2012revisión y edición, LS y AV; visualización, PL; supervisión y administración de proyectos, LS, ÉP y SF; adquisición de fondos, LS y SF. Todos los autores contribuyeron al artículo y aprobaron la versión presentada.
Financiación
Esta investigación forma parte de la Red Canadiense de Innovación Alimentaria (CFIN; ASC-14) y financiado en parte por el Gobierno de Canadá bajo el Programa AgriScience de la Asociación Agrícola Canadiense, que es una iniciativa federal, provincial y territorial. También fue financiado por el Ministère de l’Agriculture, des Pêcheries et de l’Alimentation du Québec (MAPAQ) como parte de la Chaire de recherche MAPAQ sur la qualité et la salubrité de la viande et des produits de viande transformée (Musculo; PPIA13). Sollio Cooperative Group fue socio en este proyecto.
Reconocimientos
Reconocemos al Dr. B. Boucher y al Dr. J. Brochu por sus valiosas contribuciones como veterinarios. Los autores agradecen a las plataformas del Instituto de Nutrición y Alimentos Funcionales y al Instituto de Biología y Sistemas Integrativos por su asistencia técnica.
Conflicto de intereses
Los autores AV y LS declararon que eran miembros del consejo editorial de Frontiers en el momento de la presentación. Esto no tuvo ningún impacto en el proceso de revisión por pares y la decisión final. Los autores ÉP y SF fueron empleados por la empresa Olymel S.E.C./L.P.
Los autores restantes declaran que la investigación se realizó en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un posible conflicto de intereses.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o reclamo que pueda ser hecho por su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fsysb.2023.1183868/full#supplementary-material
Referencias
Adhikari, B., Kim, S. W. y Kwon, Y. M. (2019). Caracterización de la microbiota asociada a digesta y mucosa en diferentes regiones del tracto gastrointestinal de cerdos de cría. Int. J. Mol. 20 (7), 1630. doi:10.3390/ijms20071630
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. y Lipman, D. J. (1990). Herramienta básica de búsqueda de alineación local. J. Mol. Biol. 215 (3), 403–410. doi:10.1016/S0022-2836(05)80360-2
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Andersen, K. S., Kirkegaard, R. H., Karst, S. M. y Albertsen, M. (2018). ampvis2: un paquete R para analizar y visualizar datos de amplicones de ARNr 16S. bioRxiv 1, 299537. doi:10.1101/299537
Andrews, S. (2010). FastQC: Una herramienta de control de calidad para datos de secuencia de alto rendimiento. Disponible en: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Baer, A. A., Miller, M. J. y Dilger, A. C. (2013). Patógenos de interés para la industria porcina: Una revisión de la investigación sobre intervenciones para garantizar la seguridad alimentaria. Compr. Rev. Food Sci. Food Saf. 12 (2), 183–217. doi:10.1111/1541-4337.12001
Brenner, D. J. y Farmer, J. J. (2015). Enterobacteriaceae. El hombre de Bergey. Syst. Archaea Bact. 24, 1. doi:10.1002/9781118960608.fbm00222
Bridier, A., Le Grandois, P., Moreau, M.-H., Prénom, C., Le Roux, A., Feurer, C., et al. (2019). Impacto de los procedimientos de limpieza y desinfección en la ecología microbiana y la resistencia antimicrobiana a Salmonella en un matadero de cerdos. 9 (1), 12947. doi:10.1038/s41598-019-49464-8
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Byrne, B., Lyng, J., Dunne, G. y Bolton, D. J. (2008). Una evaluación de la calidad microbiana del aire dentro de una planta de procesamiento de carne de cerdo. Control de alimentos 19 (9), 915–920. doi:10.1016/j.foodcont.2007.08.016
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A. y Holmes, S. P. (2016). DADA2: Inferencia de muestra de alta resolución a partir de datos de amplicones Illumina. Métodos 13 (7), 581–583. doi:10.1038/nmeth.3869
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Cameron, R. D. A. (2000). Una revisión de la industrialización de la producción porcina en todo el mundo, con especial referencia a la región asiática. Sanidad animal e integración en toda la zona. Brisbane, Australia: FAO.
Crespo-Piazuelo, D., Estellé, J., Revilla, M., Criado-Mesas, L., Ramayo-Caldas, Y., Óvilo, C., et al. (2018). Caracterización de las composiciones de la microbiota bacteriana a lo largo del tracto intestinal en cerdos y sus interacciones y funciones. 8 (1), 12727. doi:10.1038/s41598-018-30932-6
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. y Callahan, B. J. (2018). Identificación estadística simple y eliminación de secuencias de contaminantes en datos de genes marcadores y metagenómica. Microbioma 6 (1), 226. doi:10.1186/s40168-018-0605-2
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Dhariwal, A., Chong, J., Habib, S., King, I. L., Agellon, L. B. y Xia, J. (2017). MicrobiomeAnalyst: Una herramienta basada en la web para un análisis estadístico, visual y metaanalítico integral de datos de microbiomas. Ácidos nucleicos res. 45 (W1), W180–W188. doi:10.1093/nar/gkx295
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Duchaine, C., Grimard, Y., y Cormier, Y. (2000). Influencia del mantenimiento del edificio, factores ambientales y estaciones en los contaminantes en el aire de los edificios de confinamiento porcino. Aihaj 61 (1), 56–63. doi:10.1080/15298660008984515
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Farzan, A., Friendship, R. M., Cook, A. y Pollari, F. (2010). Presencia de Salmonella, Campylobacter, Yersinia enterocolitica, Escherichia coli O157 y Listeria monocytogenes en cerdos. Zoonosis Salud Pública 57 (6), 388–396. doi:10.1111/j.1863-2378.2009.01248.x
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Fosse, J., Oudot, N., Laroche, M., Rossero, A., Seegers, H., H. y Magras, C. (2008). Infección de lotes porcinos por cinco peligros zoonóticos bacterianos transmitidos por los alimentos: Variabilidad en la granja y en el sacrificio. Epidémiologie Santé Animale (53), 57–71.
Gill, C. O. (2000). «Chapter 5 – HACCP in primary processing: Red meat, p 81-122,» in HACCP in the meat industry (Duxford, Reino Unido: Woodhead Publishing).
Gill, C. O., y Sofos, J. N. (2005). «Chapter 11 – sources of microbial contamination at slaughtering plants», en Improving the safety of fresh meat (Cambridge, Reino Unido: Woodhead Publishing Limited), 231–243.
Gill, C. O., y Badoni, M. (2010). Efectos de la experiencia con los procedimientos de hisopado sobre el número de bacterias recuperadas de los cadáveres mediante hisopado con esponjas. J. Prot. Alimentos Prot. 73 (4), 747–751. doi:10.4315/0362-028x-73.4.747
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Gill, C. O., Badoni, M. y McGinnis, J. C. (1999). Evaluación de la adecuación de la limpieza de los equipos utilizados para el despiece de canales de vacuno. Int. J. Food Microbiol. 46 (1), 1–8. doi:10.1016/S0168-1605(98)00181-0
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Gill, C. O., y Bryant, J. (1993). La presencia de Escherichia coli, Salmonella y Campylobacter en equipos de depilación de canales de cerdo. Microbiol alimentario. 10 (4), 337–344. doi:10.1006/fmic.1993.1039
Hamscher, G., Pawelzick, H. T., Sczesny, S., Nau, H. y Hartung, J. (2003). Antibióticos en el polvo procedente de una granja de engorde de cerdos: ¿una nueva fuente de peligro para la salud de los agricultores? Entorno. Salud Perspectiva. 111 (13), 1590–1594. doi:10.1289/ehp.6288
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Ministerio de Salud del Canadá (1997). Determinación de Enterobacteriaceae, MFLP-43. Compend. Métodos 3, 1.
Health Canada (2020). Determinación del recuento de colonias aeróbicas en alimentos, MFHPB-18. Compend. Métodos anales. 2, 1.
Holman, D. B., Gzyl, K. E., Mou, K. T., Allen, H. K. y Cotter, P. D. (2021). Edad de destete y su efecto sobre el desarrollo del microbioma intestinal porcino y el resistoma. mSystems 6(6), 00682211-e100621. doi:10.1128/mSystems.00682-21
Holman, D., W Brunelle, B., Trachsel, J. y Allen, H. (2017). Metaanálisis para definir una microbiota central en el intestino porcino. mSystems 2 (3), e00004. doi:10.1128/mSystems.00004-17
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Huang, T., Zhang, M., Tong, X., Chen, J., Yan, G., Fang, S., et al. (2019). Comunidades microbianas en pulmones porcinos y su asociación con lesiones pulmonares. Microbio. Biotecnología. 12 (2), 289–304. doi:10.1111/1751-7915.13353
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Huffman, R. D. (2002). Tecnologías actuales y futuras para la descontaminación de canales y carnes frescas. Carne Sci. 62 (3), 285–294. doi:10.1016/S0309-1740(02)00120-1
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Hurd, H. S., Brudvig, J., Dickson, J., Mirceta, J., Polovinski, M., Matthews, N., et al. (2008). Impacto en la salud porcina en la contaminación de la canal y el riesgo transmitido por los alimentos humanos. Public Health Rep. 123 (3), 343–351. doi:10.1177/003335490812300314
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Hyun, H., Lee, M. S., Park, I., Ko, H. S., Yun, S., Jang, D.-H., et al. (2021). El análisis del modelo porcino de peritonitis inducida por heces revela el tropismo del microbioma sanguíneo. Frente. Celda. Infecta. Microbiol. 11, 676650. doi:10.3389/fcimb.2021.676650
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Comisión Internacional de Especificaciones Microbiológicas para Alimentos (ICMSF) (2018). «Capítulo 12 – muestreo para evaluar el control del medio ambiente», en Microorganismos en alimentos 7: Pruebas microbiológicas en la gestión de la inocuidad de los alimentos (Cham, Suiza: Springer International Publishing), 263-292.
Juni, E. (2015). Psychrobacter. Manual de Bergey de sistemática de arqueas y bacterias. Nueva York, NY: John Wiley & Sons, Inc.
Kalton, G., y Flores-Cervantes, I. (2003). Métodos de ponderación. J. estadísticas oficiales 19 (2), 81–97.
Kim, H. B. e Isaacson, R. E. (2015). La diversidad microbiana del intestino del cerdo: Comprender la ecología microbiana del intestino del cerdo a través de la secuenciación de alto rendimiento de la próxima generación. Microbiol veterinario. 177 (3), 242–251. doi:10.1016/j.vetmic.2015.03.014
Kim, J., Nguyen, S. G., Guevarra, R. B., Lee, I. y Unno, T. (2015). Análisis de la microbiota fecal porcina en diversas etapas de crecimiento. Archivos Microbiol. 197 (6), 753–759. doi:10.1007/s00203-015-1108-1
Kim, K. Y. y Ko, H. J. (2019). Características de distribución en interiores de bacterias en el aire en edificios de cerdos influenciadas por la temporada y el tipo de alojamiento. Asian-Australas J. Animal Sci. 32 (5), 742–747. doi:10.5713/ajas.18.0415
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2012). Evaluación de cebadores generales de PCR del gen de ARN ribosómico 16S para estudios de diversidad basados en secuenciación clásica y de próxima generación. Ácidos nucleicos res. 41 1), E1. doi:10.1093/nar/gks808
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Knights, D., Kuczynski, J., Charlson, E. S., Zaneveld, J., Mozer, M. C., Collman, R. G., et al. (2011). Seguimiento de fuentes microbianas independientes del cultivo en toda la comunidad bayesiana. Métodos nacionales 8 (9), 761–763. doi:10.1038/nmeth.1650
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Kraemer, J. G., Aebi, S., Oppliger, A. y Hilty, M. (2019). La microbiota del aire interior de las granjas porcinas impulsa la composición de la microbiota nasal de los criadores de cerdos de una manera dependiente de la temporada y específica de la granja. Aplicación. Entorno. Microbiol. 85 (9), E03038-18–E03018. doi:10.1128/AEM.03038-18
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Kumari, P., Woo, C., Yamamoto, N., y Choi, H.-L. (2016). Variaciones en abundancia, diversidad y composición comunitaria de hongos en el aire en casas porcinas a través de las estaciones. 6 (1), 37929. doi:10.1038/srep37929
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Le Floc’h, N., Knudsen, C., Gidenne, T., Montagne, L., Merlot, E. y Zemb, O. (2014). Impacto de la restricción alimenticia en la salud, la digestión y la microbiota fecal de cerdos en crecimiento alojados en buenas o malas condiciones de higiene. Animal 8 (10), 1632–1642. doi:10.1017/S1751731114001608
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Lebel, P., Letellier, A., Longpré, J., Laplante, B., Yergeau, E. y Fravalo, P. (2017). Las opciones de presentación de alimento en el engorde temprano porcino mitigan la excreción de Salmonella y modulan específicamente la microbiota fecal. J. Appl. Microbiol. 122 (1), 30–39. doi:10.1111/jam.13305
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Legendre, P., y Gallagher, E. D. (2001). Transformaciones ecológicamente significativas para la ordenación de datos de especies. Oecologia 129 (2), 271–280. doi:10.1007/s004420100716
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Li, Z., Hu, G., Zhu, L., Sun, Z., Jiang, Y., Gao, M.-J., et al. (2021). Estudio del crecimiento, metabolismo y morfología de Akkermansia muciniphila con un reactor intestinal biónico avanzado in vitro. BMC Microbiol. 21 (1), 61. doi:10.1186/s12866-021-02111-7
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Lin, H., y Peddada, S. D. (2020). Análisis de composiciones de microbiomas con corrección de sesgo. Nat. Commun. 11 (1), 3514. doi:10.1038/s41467-020-17041-7
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Lo Fo Wong, D. M. A., Dahl, J., Stege, H., van der Wolf, P. J., Leontides, L., von Altrock, A., et al. (2004). Factores de riesgo a nivel de rebaño para la infección subclínica por Salmonella en rebaños europeos de cerdos de engorde. Prev. Veterinaria Med. 62 (4), 253–266. doi:10.1016/j.prevetmed.2004.01.001
Longpré, J., Fairbrother, J. M., Fravalo, P., Arsenault, J., LeBel, P., Laplante, B., et al. (2016). Impacto de la alimentación con puré versus pellets en los niveles de ácido propiónico/butírico y en la carga total de Escherichia coli en el tracto gastrointestinal de cerdos en crecimiento1. J. Animal Sci. 94 (3), 1053–1063. doi:10.2527/jas.2015-9617
Lozupone, C., y Knight, R. (2005). UniFrac: Un nuevo método filogenético para comparar comunidades microbianas. Aplicación. Entorno. Microbiol. 71 (12), 8228–8235. doi:10.1128/AEM.71.12.8228-8235.2005
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Maciorowski, K. G., Herrera, P., Jones, F. T., Pillai, S. D. y Ricke, S. C. (2007). Efectos en aves de corral y ganado de la contaminación de piensos con bacterias y hongos. Alimentación Animal Sci. Technol. 133 (1), 109–136. doi:10.1016/j.anifeedsci.2006.08.006
Maes, S., Heyndrickx, M., Vackier, T., Steenackers, H., Verplaetse, A. y Reu, K. D. (2019). Identificación y potencial de deterioro de la microbiota dominante restante en superficies en contacto con alimentos después de la limpieza y desinfección en diferentes industrias alimentarias. J. Prot. Alimentos Prot. 82 (2), 262–275. doi:10.4315/0362-028X. JFP-18-226
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Mann, E., Wetzels, S. U., Pinior, B., Metzler-Zebeli, B. U., Wagner, M. y Schmitz-Esser, S. (2016). Los spoilers psiquiófilos dominan el microbioma bacteriano en muestras de musculatura de cerdos sacrificados. Carne Sci. 117, 36–40. doi:10.1016/j.meatsci.2016.02.034
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Mark Welch, J. L., Dewhirst, F. E. y Borisy, G. G. (2019). Biogeografía del microbioma oral: La hipótesis del sitio-especialista. Annu. Rev. Microbiol. 73 (1), 335–358. doi:10.1146/annurev-micro-090817-062503
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Markowiak-Kopeć, P. y Śliżewska, K. (2020). El efecto de los probióticos en la producción de ácidos grasos de cadena corta por el microbioma intestinal humano. Nutrientes 12 (4), 1107. doi:10.3390/nu12041107
Resumen de PubMed | Texto completo de CrossRef | Google Académico
McMurdie, P. J., y Holmes, S. (2013). phyloseq: Un paquete R para análisis interactivos reproducibles y gráficos de datos censales de microbiomas. PLOS ONE 8 (4), e61217. doi:10.1371/journal.pone.0061217
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Meer, Y. v. d., Gerrits, W. J. J., Jansman, A. J. M., Kemp, B. y Bolhuis, J. E. (2017). Un vínculo entre el comportamiento dañino en cerdos, las condiciones sanitarias y el suministro de proteínas y aminoácidos en la dieta. PLOS ONE 12 (5), e0174688. doi:10.1371/journal.pone.0174688
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Mendiburu, F. y Yaseen, M. (2021). agricolae: Procedimientos estadísticos para la investigación agrícola. Disponible en https://cran.r-project.org/package=agricolae.
Mikkelsen Lene, L., Naughton Patrick, J., Hedemann Mette, S. y Jensen Bent, B. (2004). Efectos de las propiedades físicas de los piensos sobre la ecología microbiana y la supervivencia de Salmonella enterica serovar Typhimurium en el tracto gastrointestinal del cerdo. Aplicación. Entorno. Microbiol. 70 (6), 3485–3492. doi:10.1128/AEM.70.6.3485-3492.2004
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Møretrø, T. y Langsrud, S. (2017). Bacterias residenciales en superficies en la industria alimentaria y sus implicaciones para la seguridad y calidad de los alimentos. Compr. Rev. Food Sci. Food Saf. 16 (5), 1022–1041. doi:10.1111/1541-4337.12283
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Mou, K. T., Allen, H. K., Alt, D. P., Trachsel, J., Hau, S. J., Coetzee, J. F., et al. (2019). Cambios en la microbiota nasal de cerdos en respuesta a diferentes regímenes de dosificación de administración de oxitetraciclina. Microbiol veterinario. 237, 108386. doi:10.1016/j.vetmic.2019.108386
Murase, K., Watanabe, T., Arai, S., Kim, H., Tohya, M., Ishida-Kuroki, K., et al. (2019). Caracterización de la saliva de cerdo como el principal hábitat natural de Streptococcus suis mediante el análisis de microbiota oral, fecal, vaginal y ambiental. PLOS ONE 14 (4), e0215983. doi:10.1371/journal.pone.0215983
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Nehme, B., Létourneau, V., Forster, R. J., Veillette, M. y Duchaine, C. (2008). Enfoque independiente del cultivo de la diversidad de bioaerosoles bacterianos en los edificios estándar de confinamiento porcino y evaluación del efecto estacional. Entorno. Microbiol. 10 (3), 665–675. doi:10.1111/j.1462-2920.2007.01489.x
Resumen de PubMed | Texto completo de CrossRef | Google Académico
O’Meara, F. M., Gardiner, G. E., O’Doherty, J. V. y Lawlor, P. G. (2020). El efecto de la forma de alimento y el método de entrega en la microbiología del alimento y el rendimiento del crecimiento en cerdos de engorde. J. animal Sci. 98 (3), skaa021. skaa021. doi:10.1093/jas/skaa021
OCDE (2021), «Meat consumption» (indicador), doi:10.1787/fa290fd0-enConsultado el 07 de enero de 2021].
Painter, J. A., Hoekstra, R. M., Ayers, T., Tauxe, R. V., Braden, C. R., Angulo, F. J., et al. (2013). Atribución de enfermedades, hospitalizaciones y muertes transmitidas por los alimentos a los productos alimenticios mediante el uso de datos de brotes, Estados Unidos, 1998-2008. Emerg. Infecta. Dis. 19 (3), 407–415. doi:10.3201/eid1903.111866
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Palleroni, N. J. (2015). Pseudomonas. Manual de Bergey de sistemática de arqueas y bacterias. Nueva York, NY: John Wiley & Sons, Inc.
Paramithiotis, S., Pappa, A. M., Drosinos, E. H. y Zoiopoulos, P. E. (2009). Parámetros microbiológicos, fisicoquímicos y de seguridad de las dietas animales basadas en cereales. Qual. Assur. Saf. Cultivos Alimentos 1 (3), 170–178. doi:10.1111/j.1757-837X.2009.00028.x
Prodan, A., Tremaroli, V., Brolin, H., Zwinderman, A. H., Nieuwdorp, M. y Levin, E. (2020). Comparación de tuberías bioinformáticas para la secuenciación de amplicones microbianos de ARNr 16S. PLOS ONE 15 (1), e0227434. doi:10.1371/journal.pone.0227434
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). Silva: Un recurso integral en línea para datos de secuencia de ARN ribosómico de calidad comprobada y alineados compatibles con ARB. Ácidos nucleicos res. 35 (21), 7188–7196. doi:10.1093/nar/gkm864
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Puertollano, E., Kolida, S. y Yaqoob, P. (2014). Importancia biológica del metabolismo de ácidos grasos de cadena corta por el microbioma intestinal. Curr. Opinar. Clin. Nutr. Atención metabólica 17 (2), 139–144. doi:10.1097/mco.0000000000000025
Quan, J., Cai, G., Ye, J., Yang, M., Ding, R., Wang, X., et al. (2018). Una comparación global de las composiciones del microbioma de tres ubicaciones intestinales en cerdos comerciales con índices extremos de conversión alimenticia. 8 (1), 4536. doi:10.1038/s41598-018-22692-0
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Ramsay, D., y Delisle, M-F. (2017). Bilan annuel 2014-2015 – toxi-infecciones alimentaires. Québec. Disponible en: https://www.mapaq.gouv.qc.ca/fr/Publications/Bilan_Toxi-infection_2014-2015_Accessible.pdf (consultado el 08 de mayo de 2019).
Remenant, B., Jaffrès, E., Dousset, X., Pilet, M.-F. y Zagorec, M. (2015). Spoilers bacterianos de los alimentos: comportamiento, estado físico y propiedades funcionales. Microbiol alimentario. 45, 45–53. doi:10.1016/j.fm.2014.03.009
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Roussel, C., Chabaud, S., Lessard-Lord, J., Cattero, V., Pellerin, F. A., Feutry, P., et al. (2022). La virulencia colónica y la urovirulencia de UPEC son mitigadas por metabolitos microbianos de extracto de arándano rico en proantocianidinas en un modelo intestinal y un urotelio de ingeniería tisular en 3D. Microbiol. Espectro. 10 (5), E0243221. doi:10.1128/spectrum.02432-21
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Ruttkies, C., Schymanski, E. L., Wolf, S., Hollender, J. y Neumann, S. (2016). Relanzamiento de MetFrag: Incorporando estrategias más allá de la fragmentación in silico. J. Cheminformatics 8 (1), 3. doi:10.1186/s13321-016-0115-9
Saucier, L. (2016). Deterioro microbiano, calidad y seguridad en el contexto de la sostenibilidad de la carne. Carne Sci. 120, 78–84. doi:10.1016/j.meatsci.2016.04.027
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Schliep, K. P. (2010). phangorn: análisis filogenético en R. Bioinformatics 27 (4), 592–593. doi:10.1093/bioinformatics/btq706
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Descubrimiento y explicación de biomarcadores metagenómicos. Genoma Biol. 12 (6), R60. doi:10.1186/gb-2011-12-6-r60
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Song, L., Wang, C., Jiang, G., Ma, J., Li, Y., Chen, H., et al. (2021). El bioaerosol es una importante vía de transmisión de genes de resistencia a antibióticos en granjas porcinas. Entorno Int. 154, 106559. doi:10.1016/j.envint.2021.106559
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Spescha, C., Stephan, R. y Zweifel, C. (2006). Contaminación microbiológica de canales de cerdo en diferentes fases de sacrificio en dos mataderos autorizados por la Unión Europea. J. Prot. Alimentos Prot. 69 (11), 2568–2575. doi:10.4315/0362-028X-69.11.2568
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Stärk, K., Miserez, R., Siegmann, S., Ochs, H., Infanger, P. y Schmidt, J. (2008). Erradicación de las enfermedades respiratorias enzoóticas de los cerdos mediante un programa de control nacional en suiza: -ES- Un exitoso programa nacional de control de enfermedades respiratorias enzoóticas en cerdos en Suiza -fr- un programme réussi de lutte contre les maladies respiratoires enzootiques des porcs en Suisse -es-. Revue Sci. Tech. Int. Office Epizootias) 26, 595–606. doi:10.20506/rst.26.3.1768
Vigors, S., O’ Doherty, J. V. y Sweeney, T. (2020). Los perfiles del microbioma colónico para mejorar la eficiencia alimenticia se pueden identificar a pesar de los principales efectos de la granja de origen y el grupo contemporáneo en cerdos. Animal 14 (12), 2472–2480. doi:10.1017/S1751731120001500
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Wang, M., Wichienchot, S., He, X., Fu, X., Huang, Q. y Zhang, B. (2019). Fermentación colónica in vitro de fibras dietéticas: tasa de fermentación, producción de ácidos grasos de cadena corta y cambios en la microbiota. Tendencias Food Sci. Technol. 88, 1–9. doi:10.1016/j.tifs.2019.03.005
Wang, Q., Cai, R., Huang, A., Wang, X., Qu, W., Shi, L., et al. (2018). Comparación de la microbiota orofaríngea en lechones sanos y lechones con enfermedad respiratoria. Frente. Microbiol. 9, 3218. doi:10.3389/fmicb.2018.03218
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Wang, X., Li, X., Zhao, C., Hu, P., Chen, H., Liu, Z., et al. (2012). Correlación entre la composición de la comunidad bacteriana y la concentración de ácidos grasos volátiles en el rumen durante el período de transición y la cetosis en vacas lecheras. Aplicación. Entorno. Microbiol. 78 (7), 2386–2392. doi:10.1128/AEM.07545-11
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Warriner, K., Aldsworth, T. G., Kaur, S. y Dodd, C. E. R. (2002). Contaminación cruzada de canales y equipos durante el procesamiento de carne de cerdo. J. Appl. Microbiol. 93 (1), 169–177. doi:10.1046/j.1365-2672.2002.01678.x
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Wheatley, P., Giotis, E. S. y McKevitt, A. I. (2014). Efectos de las operaciones de sacrificio sobre la contaminación de la canal en una planta de producción de carne de cerdo irlandesa. Ir. Veterinario J. 67 (1), 1. doi:10.1186/2046-0481-67-1
Wright, E. S. (2016). Uso de DECIPHER v2.0 para analizar grandes datos de secuencias biológicas en R. R J. 8, 352–359. doi:10.32614/rj-2016-025
Xiao, Y., Kong, F., Xiang, Y., Zhou, W., Wang, J., Yang, H., et al. (2018). Biogeografía comparativa del microbioma intestinal entre cerdos Jinhua y Landrace. 8 (1), 5985. doi:10.1038/s41598-018-24289-z
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Yan, H., Li, Y., Zhang, Y., Zhang, H., Guo, Z. y Liu, J. (2021). Desciframiento de la diversidad microbiana y el resistoma antibiótico de bioaerosoles en edificios de confinamiento porcino. Sci. Entorno total. 781, 147056. doi:10.1016/j.scitotenv.2021.147056
Zagorec, M., y Champomier-Vergès, M-C. (2017). «Capítulo 6 – microbiología de la carne y deterioro», en Lawrie’s meat science (Duxford, Reino Unido: Woodhead Publishing), 187–203.
Zhao, W., Wang, Y., Liu, S., Huang, J., Zhai, Z., He, C., et al. (2015). La distribución dinámica de la microbiota porcina en diferentes edades y segmentos del tracto gastrointestinal. PLOS ONE 10 (2), e0117441. doi:10.1371/journal.pone.0117441
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zweifel, C. y Stephan, R. (2014). «Capítulo 16 – contaminación microbiana durante el sacrificio», en Inspección y control de carne en el matadero (Hoboken, Estados Unidos: John Wiley & Sons), 423–438.
Zwirzitz, B., Wetzels, S. U., Dixon, E. D., Stessl, B., Zaiser, A., Rabanser, I., et al. (2020). Las fuentes y rutas de transmisión de las poblaciones microbianas a través de una instalación de procesamiento de carne. npj Biofilms Microbiomas 6 (1), 26. doi:10.1038/s41522-020-0136-z
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zwirzitz, B., Wetzels, S. U., Rabanser, I., Thalguter, S., Dzieciol, M., Wagner, M., et al. (2019). Evaluación independiente del cultivo de patrones de contaminación bacteriana en canales de cerdo en una instalación de sacrificio comercial. J. Prot. Alimentos Prot. 82 (10), 1677–1682. doi:10.4315/0362-028X. JFP-19-103
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Cita: Laforge P, Vincent AT, Duchaine C, Feutry P, Dion-Fortier A, Plante P-L, Pouliot É, Fournaise S y Saucier L (2023) Contribución de las granjas a la microbiota en la cadena de valor porcina. Frente. Sistema Biol. 3:1183868. doi: 10.3389/fsysb.2023.1183868
Recibido: 10 de marzo de 2023; Aprobado: 15 de junio de 2023;
Publicado: 12 julio 2023.
Editado por:
Helena U. Zacharias, Escuela de Medicina de Hannover, Alemania
Revisado por:
Bouabid Badaoui, Universidad Mohammed V, Marruecos
Chrysovalantou Chatziioannou, Agencia Internacional para la Investigación del Cáncer (IARC), Francia
Hassan Zafar, Instituto Centroeuropeo de Tecnología (CEITEC), Chequia
Wen Chyin Yew, Universidad de Northumbria, Reino Unido
Derechos de autor © 2023 Laforge, Vincent, Duchaine, Feutry, Dion-Fortier, Plante, Pouliot, Fournaise y Saucier. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia de Atribución Creative Commons (CC BY).
*Correspondencia: Linda Saucier, linda.saucier@fsaa.ulaval.ca
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o reclamo que pueda ser hecho por su fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía