
Primer informe de GI.1aP-GI.2 recombinantes del virus de la enfermedad hemorrágica del conejo

Primer informe de GI.1aP-GI.2 recombinantes del virus de la enfermedad hemorrágica del conejo en conejos domésticos en China
 Yan Li1,
Yan Li1,  Deyan Du2, Long Zhou1, Liyin Su1, Chengcheng Tú1, Huai Zhang1, Jifeng Yu3, Lu Xiao3 y
Deyan Du2, Long Zhou1, Liyin Su1, Chengcheng Tú1, Huai Zhang1, Jifeng Yu3, Lu Xiao3 y  Jian Huang1*
Jian Huang1*- 1Facultad de Ciencia Animal y Medicina Veterinaria, Southwest Minzu University, Chengdu, China
- 2Huapai Biological Group, Chengdu, China
- 3Academia de Ciencia Animal de Sichuan, Laboratorio Provincial Clave de Cría de Animales y Genética de Sichuan, Chengdu, China
El virus de la enfermedad hemorrágica del conejo 2 (RHDV2 o GI.2) es un agente altamente contagioso que conduce a la enfermedad letal en conejos. Con frecuencia se recombina con otros géneros de Lagovirus, generando variantes epidémicas con alta patogenicidad. En este estudio, veintidós muestras de hígado dieron positivo para el gen GI.2 VP60, se recolectaron en granjas de conejos de varias regiones geográficas de China. Todas las muestras GI.2 positivas se sometieron a detección por RT-PCR, secuenciación de nucleótidos y análisis filogenético. Además, se evaluó la virulencia del virus de los recombinantes GI.2 sospechosos. Los resultados mostraron que nueve presuntos recombinantes fueron identificados mediante pruebas de recombinación RdRp-VP60. En estos recombinantes, se seleccionaron cuatro para caracterizar completamente el genoma de las nuevas variantes recombinantes GI.2, que se describieron como GI.1aP-GI.2. La secuencia de nucleótidos de estas nuevas variantes mostró un patrón de recombinación único y características filogenéticas en comparación con las variantes GI.2 actualmente prevalentes. Además, esta recombinación distintiva de la nueva variante SCNJ-2021 mejoró moderadamente la virulencia de GI.2, incluso para conejos vacunados contra GI.2 parental. En conclusión, los nuevos recombinantes GI.1aP-GI.2 se identificaron por primera vez en la industria del conejo en China, lo que amplió el conocimiento sobre la filodinámica y la diversidad genómica del genotipo GI.2. La rápida evolución molecular y la variada patogenicidad de estos virus recombinantes ponen de relieve la necesidad urgente de vigilancia epidemiológica y de prevención futura de estas variantes GI.2 desatendidas.
1. Introducción
El virus de la enfermedad hemorrágica del conejo (RHDV) es un agente común y altamente contagioso que causa el síndrome hemorrágico multiorgánico agudo con alta morbilidad y mortalidad (Teifke et al., 2002; Neimanis et al., 2018a). El RHDV es un virus de ARN monocatenario de sentido positivo del género Lagovirus, familia Caliciviridae (Lopes et al., 2017). Cada partícula contiene un genoma de aproximadamente 7.4 kb y un ARN subgenómico (sgRNA) de aproximadamente 2.5 kb. El genoma codifica dos marcos de lectura abiertos (ORF) con una ligera superposición. ORF1 codifica una poliproteína grande que es escindida por una proteasa codificada por virus, generando siete proteínas no estructurales (NSP) y la proteína estructural principal (VP60), y ORF2 codifica una proteína estructural menor (VP10) (Le Pendu et al., 2017). Con base en la clasificación filogenética y la variabilidad del gen VP60, los RHDV se dividieron en genotipos GI.1 y GI.2. El genotipo GI.1 se subdividió en RHDV clásico (G1 / GI.1b, G2 / GI.1c y G3-G5 / GI.1d) y variante antigénica RHDVa (G6 / GI.1a) (Le Pendu et al., 2017).
Desde el primer brote en la provincia de Jiangsu, China, en 1984 (Liu et al., 1984), los RHDV clásicos que experimentaron una evolución constante con alteraciones genómicas acumulativas presentaron características etiológicas y epidemiológicas variadas (Wang et al., 2012; Abrantes et al., 2020a). En los últimos veinte años, los genotipos GI.1c y GI.1a circularon conjuntamente en China junto con la recombinación intergenotípica durante su transmisión generalizada (Hu et al., 2016, 2017). En 2010, se identificó una nueva variante del RHDV en Francia, llamada RHDVb o RHDV2 (GI.2), que mostró características genéticas y antigénicas distintivas en comparación con GI.1. Además, este virus exhibió una baja protección cruzada con otros lagovirus (Le Gall-Reculé et al., 2011). Posteriormente, el GI.2 altamente patógeno dañó fuertemente la industria del conejo en Europa, Australia, África y América del Norte, que rápidamente reemplazó a GI.1 como genotipo predominante en la última década (Dalton et al., 2012; Mahar et al., 2018; Neimanis et al., 2018b; Chehida y otros, 2021; Aguayo-Adán et al., 2022). Debido a la rápida propagación de los RHDV y las pérdidas económicas y ecológicas resultantes, los GI.1a y GI.2 patógenos emergentes plantearon una mayor preocupación en los últimos años (Lopes et al., 2017; Pacioni et al., 2022). Estas circunstancias pueden implicar un conocimiento continuo de la diversidad genómica y la alteración de la virulencia de GI.2 como consecuencia de su transmisión persistente en conejos.
En 2020, la cepa GI.2 se identificó en la provincia de Sichuan en China (Hu et al., 2021), durante un brote de RHD, que se sospechaba que era el resultado de la importación internacional debido a su alta homología de nucleótidos con aislados de los Países Bajos en 2016 (Qi et al., 2022). Recientemente, la recombinación intergenotípica entre segmentos del genoma no estructurales y estructurales derivados de diferentes genotipos se consideró como el principal mecanismo de evolución genética en Lagovirues (Mahar et al., 2021). Por lo tanto, este mecanismo también puede ser un controlador robusto para las variantes GI.2 para ampliar el rango de host y la adaptación. Hasta ahora, se han confirmado varios patrones de recombinación para las variantes GI.2, incluida la recombinación intergenotípica entre GI.1b y GI.2 patógenos (por ejemplo, GI.1bP-GI.2), entre RCV no patógeno y GI.2 (por ejemplo, GI.4eP-GI.2, GI.4 cP-GI.2, GI.3P-GI.2) (Lopes et al., 2017; Mahar et al., 2018; Silvério et al., 2018; Abrantes et al., 2020b). Las variantes GI.4eP-GI.2 y GI.4 cP-GI.2 están reemplazando progresivamente a la GI.2 parental anterior en un período relativamente corto, fortaleciendo la inferencia de que la sustitución del genoma en la región no estructural puede acelerar la adaptación evolutiva del virus (Mahar et al., 2021) y alterar su virulencia (Smertina et al., 2021).
Para comprender la dinámica de GI.2 en conejos domésticos desde su invasión en China continental, caracterizamos el genoma de las variantes GI.2 de preocupación y confirmamos su alteración patogénica en el presente estudio. Aquí, primero describimos eventos de recombinación entre GI.1a y GI.2 en granjas de conejos en China, que generaron las nuevas variantes GI.1aP-GI.2. Los resultados de este estudio enfatizan la necesidad de implementar la vigilancia epidemiológica de los lagovirus para desentrañar su cocirculación y evolución, con el fin de adaptar el programa de prevención.
2. Materiales y métodos
2.1. Recogida de muestras y detección molecular
Se recolectaron veintidós muestras de hígado de conejo de doce granjas de conejos afectadas por RHD en las provincias de Sichuan, Shandong, Anhui y Yunnan, desde mayo de 2020 hasta noviembre de 2022. El ARN total se extrajo de las muestras de hígado utilizando el reactivo RNAiso plus (TaKaRa, China), luego se realizó la transcripción inversa con el PrimeScript™ RT Reagent Kit (TaKaRa, China). Todas las muestras se confirmaron como GI.2 positivas mediante un ensayo diferencial Taqman RT-PCR como se describió anteriormente (Zhou et al., 2022).
Los cebadores dirigidos a la región RdRp-VP60 se diseñaron utilizando el software Primer Premier 6.0 (PREMIER Biosoft, EE. UU.) para generar un amplicón de 994 pb de largo mediante RT-PCR, luego los productos de PCR se secuenciaron utilizando la plataforma ABI 3730XL (Sangon Biotech Co., China) para un análisis de recombinación adicional. Se utilizaron ocho pares de cebadores que abarcan el genoma GI.2 completo para obtener los productos de PCR de cinco cepas GI.2 representativas. Los productos de PCR se purificaron e insertaron en el vector pMD19-T (TaKaRa, China), y al menos tres clones positivos de cada fragmento se enviaron para la secuenciación de ácidos nucleicos. La información sobre todos los cebadores y muestras clínicas se enumeró en la Tabla Suplementaria S1, S2.
2.2. Histopatología y microscopía electrónica de transmisión
Se registraron los hallazgos patológicos macroscópicos en los conejos muertos, y se realizaron necropsias posteriores de acuerdo con los procedimientos de rutina. Los bloqueos de tejido hepático se fijaron en paraformaldehído al 4% durante 24 h, luego se incrustaron en parafina, se seccionaron a 4 μm y se tiñeron con hematoxilina y eosina. La histopatología de la sección hepática se observó bajo el microscopio óptico (Leica, Alemania). Para la microscopía electrónica de transmisión (TEM), las partículas de virus se purificaron como se describió anteriormente (Hu et al., 2010) con una modificación menor. Los tejidos hepáticos infectados se homogeneizaron y congelaron y descongelaron rápidamente para liberar las partículas del virus. La suspensión del virus se recogió después de la centrifugación (10.000 g, 20 min) a 4 °C. Luego, el sobrenadante se trató con 6% (p/v) de polietilenglicol (PEG 6000) y 3% (p/v) de NaCl durante la noche a 4°C. El precipitado se resuspendió en PBS después de una centrifugación a baja velocidad (4.450 g, 40 min, 4 °C), y luego se combinó con una mezcla de butanol e isopentanol (24:1, v/v) y se agitó durante 5 min. La suspensión se aclaró mediante centrifugación a baja velocidad (430 g, 40 min, 4 °C). La fase acuosa se recogió y centrifuga a 15.000 g durante 40 min. El sobrenadante fue enviado al centro experimental de biomedicina Chengdu Lilai para la detección de partículas de virus bajo el TEM (JEOL, Japón).
2.3. Alineación genómica y análisis filogenético
Todas las secuencias fueron recuperadas de la base de datos GenBank, incluyendo las secuencias genómicas representativas de 61 Lagovirus de diferentes genotipos (Tabla Suplementaria S3). Las secuencias genómicas completas de SCMS-2020 (acceso a GenBank: OQ570964), SCNJ-2021 (acceso a GenBank: OQ570963), SDRZ-2021 (acceso a GenBank: OQ570961), SCMS-2022 (acceso a GenBank: OQ570960) y AHFY-2022 (acceso a GenBank: OQ570962) se obtuvieron mediante ensamblaje de secuencias. La identidad de nucleótidos y aminoácidos de la alineación de cepas RHDV se analizó utilizando el programa MegAlign dentro del software DNASTAR 7.0 (DNASTAR Inc., Madison, WI, EUA). El análisis filogenético de secuencias genómicas completas se realizó utilizando MEGA 10 con el enfoque de máxima verosimilitud basado en fragmentos de NSP (posiciones nt 10-5304), fragmentos VP60 (posiciones nt 5305-7044) y genoma completo utilizando el modelo GTR + G + I. La fiabilidad de los nodos se evaluó mediante el procedimiento de remuestreo bootstrap que consta de 1.000 réplicas.
2.4. Análisis de recombinación
El Programa de Detección de Recombinación 4 (RDP4, v4.24) que contiene siete algoritmos de evaluación (RDP, Bootscan, GENECONV, MaxChi, Quimera, SiScan y 3Seq) se utilizó para confirmar los supuestos eventos de recombinación y los puntos de interrupción de recombinación precisos. Los eventos de recombinación se consideraron significativos (valor de p ≤1× 10−6) cuando sea compatible con al menos cinco de los siete algoritmos. SimPlot (v3.5.1, Baltimore, MD, EUA) con una ventana de 200 pb deslizándose a lo largo del genoma (tamaño de paso de 20 pb) se utilizó para analizar las nuevas variantes. Los lagovirus recombinantes se definieron utilizando la nomenclatura [genotipo RdRp]P-[genotipo de cápside].
2.5. Ensayos de hemaglutinación e inhibición de la hemaglutinación
La hemaglutinación (HA) y la inhibición de la hemaglutinación (HI) se realizaron como se describió anteriormente (Mizoguchi et al., 2003; Song et al., 2017). Para el AH, el tejido hepático se homogeneizó en hielo, luego se recolectó el sobrenadante después de la centrifugación. Los glóbulos rojos humanos tipo B se lavaron en solución salina tamponada con fosfato (PBS) y luego se centrifugaron (280 g, 10 min) a temperatura ambiente. Los pellets de glóbulos rojos se resuspendieron y diluyeron en PBS (pH 7.2) hasta la concentración final del 1%. Luego, se agregaron sobrenadantes de 25 μL de homogeneizado hepático en placas de microtitulación inferior en forma de V de 96 pocillos y se diluyó en serie dos veces con el mismo volumen de PBS (pH 7.2). Más tarde, se agregaron 25 μL de glóbulos rojos humanos tipo B al 1% a cada pocillo y se incubaron a 25 ° C durante 30-60 min. El título de HA se determinó como la dilución más alta que causó hemaglutinación completa de los glóbulos rojos.
Para el HI, los sueros recolectados fueron inactivados y pretratados con caolín al 25% (Macklin, China). Luego, se agregaron 25 μL de suero en placas de microtitulación inferior en forma de V de 96 pocillos y se diluyó en serie dos veces con el mismo volumen de PBS 25 μL de antígenos RHDV (4 HAU) en cada pocillo y se incubó a 25 ° C durante 30-60 min. Posteriormente, se agregaron 25 μL de glóbulos rojos humanos tipo B en cada pocillo y se establecieron a 25 ° C durante 30-60 min. La dilución más alta que causó la inhibición completa se consideró el título de inhibición de la hemaglutinación. HI título ≤23 se consideró como anticuerpo negativo.
2.6. Inoculación de la vacuna y desafío letal con GI.1aP-GI.2 en conejos experimentales
Conejos blancos de Nueva Zelanda de cuatro semanas de edad (juveniles) y de tres meses (adultos) fueron criados en el Centro de Animales Experimentales de Huapai Biological Group Co., Ltd. (Chengdu, China). Estos conejos fueron probados y demostraron ser seronegativos a GI.1/GI.2 (título HI≤ 23). Después de la alimentación adaptativa durante una semana, 60 conejos fueron asignados aleatoriamente al grupo no vacunado y al grupo vacunado (recibiendo una dosis única de vacunación bivalente inactivada contra el RHDV que consiste en el antígeno GI.1a y GI.2 inactivado). La vacuna bivalente inactivada contra el VRHD se preparó de la siguiente manera. Primero, los homogeneizados hepáticos se prepararon a partir de conejos ingenuos que murieron de RHD con desafío GI.1a o GI.2. Después de ser inactivados por formaldehído, los homogeneizados hepáticos se mezclaron con una proporción 1:1 de cada antígeno (512 HAU de cada genotipo). Los conejos en el grupo vacunado recibieron una inoculación subcutánea de 1 ml de vacuna bivalente producida en laboratorio. Para determinar la dosis provocativa del virus, se implementó el cálculo de la mediana de la dosis letal (LD50) inoculando conejos con una serie de diluciones de 10 veces de homogeneizados hepáticos que contienen la cepa SCMS-2020 o la cepa SCNJ-2021 (5 conejos por dilución). Entonces, el LD50 los valores se calcularon por el método Reed-Muench (Tabla Suplementaria S4). El LD50 los valores para la cepa SCMS-2020 y la cepa SCNJ-2021 se determinaron como 10−4,68 LD50 y 10−5,5 LD50 en homogeneizados hepáticos de 1 ml, por separado. En el día 14 después de la vacunación, los conejos en el grupo vacunado (Los títulos de anticuerpos HI estaban entre 27y 29) fueron desafiados con una dosis de 10.000 LD50 para GI.2 (cepa SCMS-2020) o 10.000 LD50 para GI.1aP-GI.2 (cepa SCNJ-2021), respectivamente. Los conejos en el grupo no vacunado también fueron desafiados con una dosis de 10,000 LD50 para cada cepa. El tiempo de supervivencia y la mortalidad se registraron dentro de las 96 h posteriores a la infección.
3. Resultados
3.1. Hallazgos patológicos e identificación de partículas de virus
Los conejos enfermos sucumbieron a la RHD fueron examinados en la necropsia y evaluados histológicamente. Los resultados mostraron que la epistaxis se observó en aproximadamente el 6% de los conejos sometidos a RHD aguda y subaguda (Figura 1A). Los lóbulos hepáticos agrandados, amarillo-marrón y moteados fueron notablemente anormales, y también se observó coagulopatía multifocal en pulmones y otros órganos (Figura 1B) en la necropsia. Se confirmaron lesiones histopatológicas significativas en el hígado de todos los animales. Necrosis celular evidente con hemorragia apareció en todo el parénquima hepático desordenado, que fue infiltrado por un gran número de heterófilos (Figura 1C). Además, la evidencia de viriones en los tejidos hepáticos infectados fue confirmada por el TEM. La simetría icosaédrica visible de las partículas del virus, de aproximadamente 30 nm de diámetro con una capa interna, fue consistente con GI.1/GI.2 (Figura 1D).
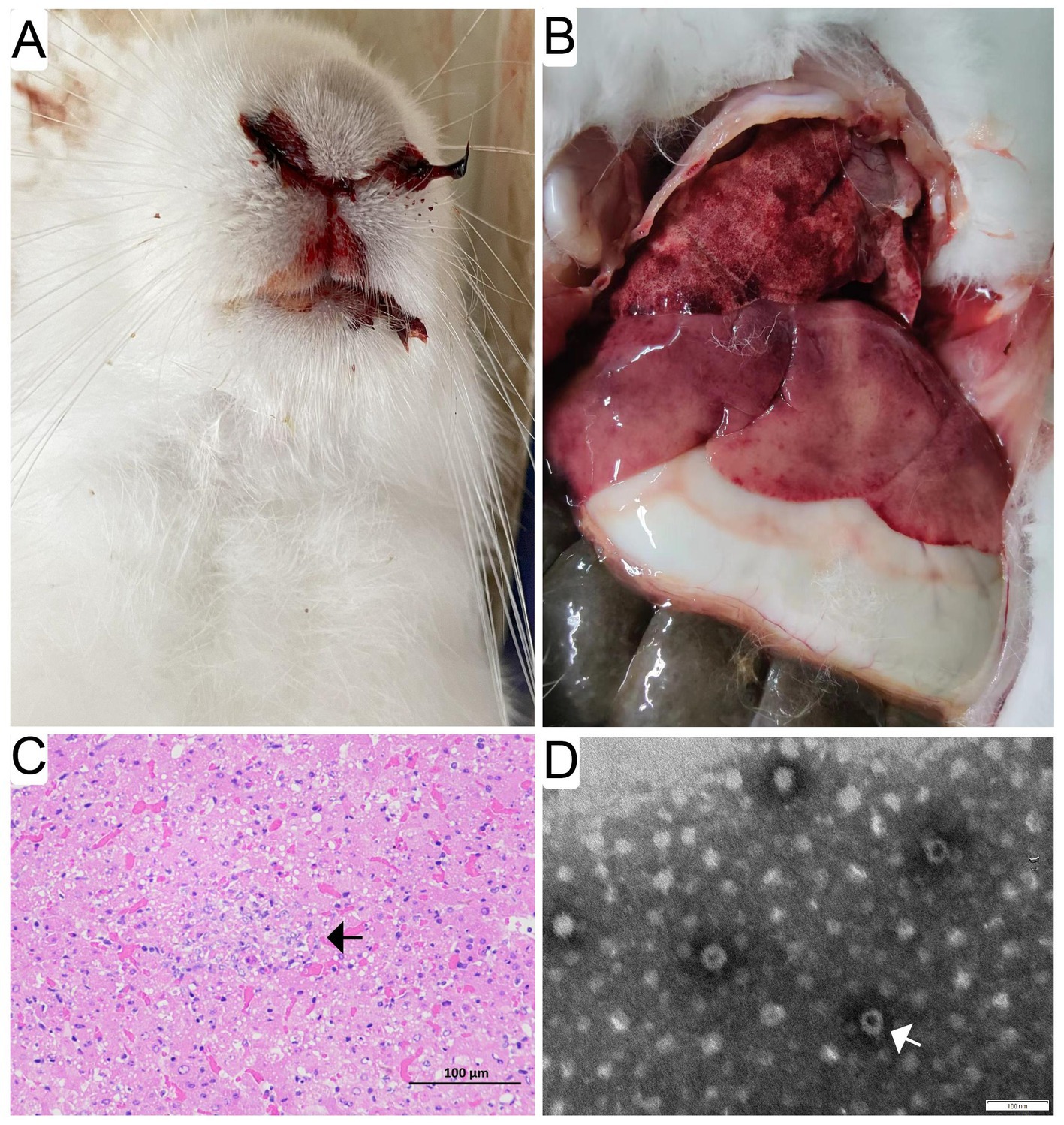 Figura 1. Hallazgos patológicos e histopatológicos macroscópicos en conejos infectados. Se observó epistaxis evidente en aproximadamente el 6% de los conejos infectados sometidos a RHD aguda y subaguda (A). Los lóbulos hepáticos agrandados, amarillo-marrón y moteados fueron notablemente anormales, y también se encontró coagulopatía multifocal en los pulmones y otros órganos (B) en la necropsia. Se confirmaron lesiones histopatológicas significativas en el hígado y el bazo de todos los animales. Evidente necrosis celular con hemorragia apareció en todo el parénquima hepático dispuesto, que fue infiltrado por un gran número de heterófilos (flecha blanca) (barra = 100 μm) (C). Las partículas de virus purificadas y teñidas negativamente se mostraron (flecha blanca) bajo micrografías electrónicas (barra = 100 nm) (D).
Figura 1. Hallazgos patológicos e histopatológicos macroscópicos en conejos infectados. Se observó epistaxis evidente en aproximadamente el 6% de los conejos infectados sometidos a RHD aguda y subaguda (A). Los lóbulos hepáticos agrandados, amarillo-marrón y moteados fueron notablemente anormales, y también se encontró coagulopatía multifocal en los pulmones y otros órganos (B) en la necropsia. Se confirmaron lesiones histopatológicas significativas en el hígado y el bazo de todos los animales. Evidente necrosis celular con hemorragia apareció en todo el parénquima hepático dispuesto, que fue infiltrado por un gran número de heterófilos (flecha blanca) (barra = 100 μm) (C). Las partículas de virus purificadas y teñidas negativamente se mostraron (flecha blanca) bajo micrografías electrónicas (barra = 100 nm) (D).
3.2. Identificación de nuevas variantes GI.1aP-GI.2
Veintidós muestras de hígado de conejo dieron positivo para el gen GI.2 VP60, se recolectaron en granjas de conejos de las provincias de Sichuan, Anhui, Shandong y Yunnan de 2020 a 2022 durante los brotes de RHD (Tabla suplementaria S2) y ninguna fue positiva para el gen GI.1 VP60. Luego, obtuvimos las secuencias de nucleótidos de la unión RdRp-VP60 derivadas de aislados GI.2 anteriores. El análisis recombinante de las uniones RdRp-VP60 confirmó que nueve de los 22 aislamientos (40,9%) eran cepas GI.1aP-GI.2 presuntamente recombinantes, mientras que las otras pertenecían a las cepas GI.2 parentales (Figura suplementaria S1; Cuadro suplementario S2). Se obtuvieron y secuenciaron ocho fragmentos superpuestos de cada cepa representativa (Figura suplementaria S2). Las alineaciones de nucleótidos de las secuencias de consenso confirmaron que SCNJ-2021 (OQ570963), SDRZ-2021 (OQ570961), SCMS-2022 (OQ570960) y AHFY-2022 (OQ570962) eran las cepas recombinantes pertenecientes al clado GI.1aP-GI.2, mientras que SCMS-2020 (OQ570964) se clasificó como un prototipo de GI.2.
3.3. Características filogenéticas de las variantes GI.aP-GI.2
Para revelar las caracterizaciones genéticas de estas variantes GI.2 recombinantes, sus secuencias genómicas se analizaron exhaustivamente mediante herramientas bioinformáticas. Los árboles filogenéticos de ML basados en la región codificante de NSP (nt 10-5304, que indica la secuencia utilizada como referencia de estas posiciones), el gen VP60 (nt 5305-7044, que indica la secuencia utilizada como referencia de estas posiciones) y el genoma completo se construyeron por separado. El análisis genético basado en los genes NSP reveló que SCNJ-2021, SDRZ-2021, SCMS-2022 y AHFY-2022 tenían 86.4-89.1% de identidad de nucleótidos y 96.0-97.8% de identidad de aminoácidos con cepas de referencia RHDVa (es decir, Triptis, Iowa2000, JX / CHA / 97) y agrupadas en una nueva rama con una cepa de virus recientemente reportada (JS-NATF2 / China / OM451150) identificada en Oryctolagus cuniculus (Figura 2A). Sin embargo, los perfiles filogenéticos basados en el gen VP60 mostraron que estas cuatro cepas recombinantes se agrupaban estrechamente con otras cepas GI.2 de referencia de China (es decir, CHN/SC2020, SC2020/0401, SC20-01 y SC-1), y la identidad de nucleótidos era de hasta 98,6-99,0% (Figura 2B). Significativamente, las cuatro variantes (SCNJ-2021, SDRZ-2021, AHFY-2022 y SCMS-2022) se ramificaron en un grupo monofilético que muestra una identidad de nucleótidos del 84.7-88.6% con otras cepas representativas basadas en el análisis completo del genoma (Figura 2C; Cuadros suplementarios S3, S6).
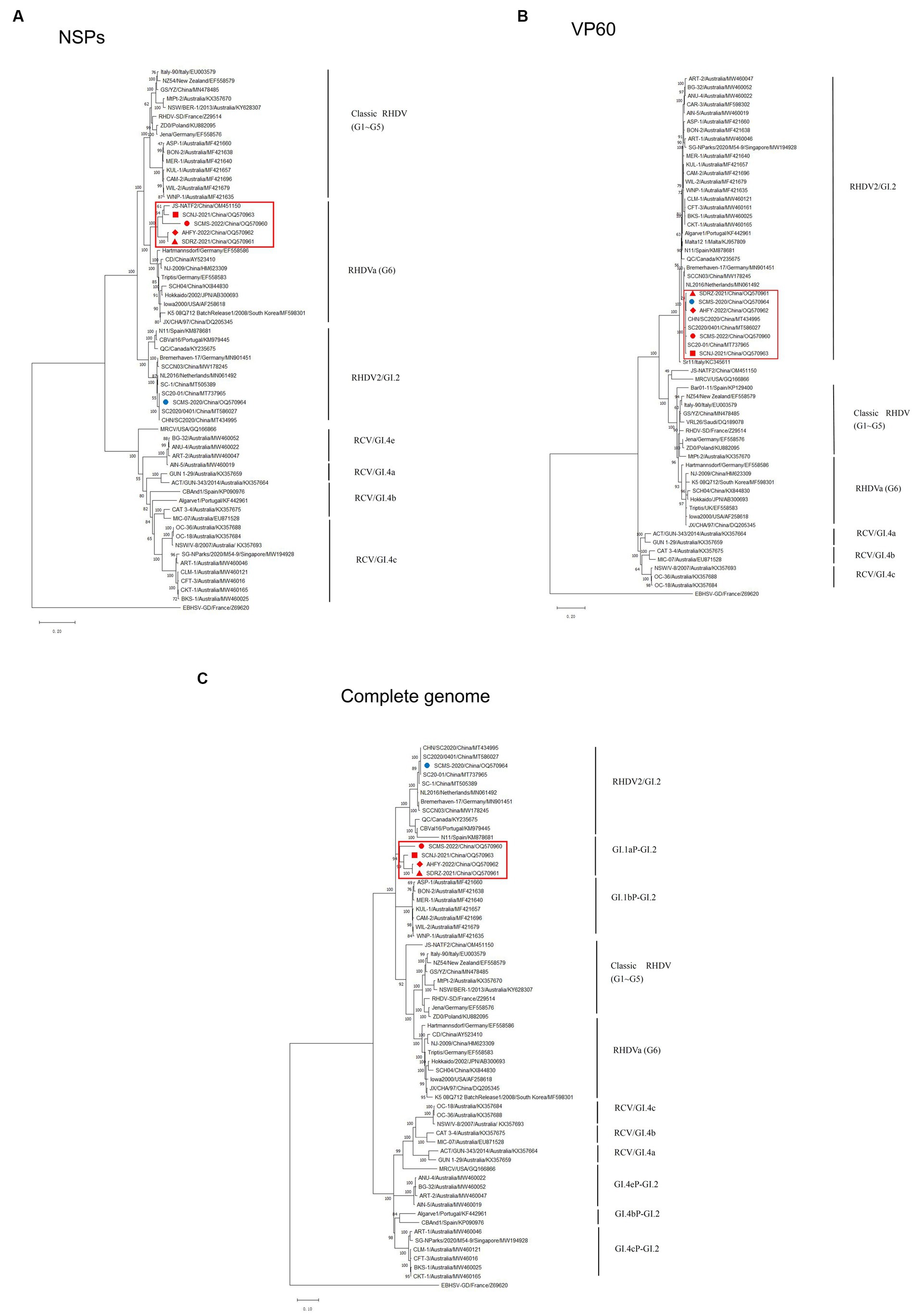 Figura 2. Árboles filogenéticos basados en NSPs (A), VP60 (B) y secuencia genómica completa (C) de cinco aislados con 56 cepas representativas de RHDV y RCV disponibles en GenBank. Se indican los principales grupos genéticos (genogrupos) y se etiquetan los cinco aislados de este estudio. El árbol filogenético se construyó utilizando el método ML (1.000 bootstrap) en MEGA 10. Los números a lo largo de las ramas son valores de arranque. La barra de escala indica el sustituto de nucleótido por sitio.
Figura 2. Árboles filogenéticos basados en NSPs (A), VP60 (B) y secuencia genómica completa (C) de cinco aislados con 56 cepas representativas de RHDV y RCV disponibles en GenBank. Se indican los principales grupos genéticos (genogrupos) y se etiquetan los cinco aislados de este estudio. El árbol filogenético se construyó utilizando el método ML (1.000 bootstrap) en MEGA 10. Los números a lo largo de las ramas son valores de arranque. La barra de escala indica el sustituto de nucleótido por sitio.
3.4. Eventos de recombinación de las variantes GI.1aP-GI.2
Para verificar aún más este nuevo patrón de recombinación, los eventos de recombinación de estas cuatro variantes fueron analizados por el Programa de Detección de Recombinación 4 y el software Simplot. Se utilizaron al menos cinco métodos para confirmar la recombinación del genoma completo de las variantes SCNJ-2021, SDRZ-2021, AHFY-2022 y SCMS-2022 mediante el análisis (valores de p de ≤1 × 10−6) (Cuadro suplementario S7). El análisis de la gráfica de similitud confirmó los puntos de ruptura de recombinación a lo largo de la unión genómica RdRp-VP60 (nt 5240, nt 5274 o nt 5304) (Figura 3). Determinadas por el software RDP4, las variantes parentales más probables para las cuatro cepas fueron GI.1a patógena que donó el segmento del genoma no estructural (accesión de Genbank EF558583) y GI.2 patógena que donó el segmento del genoma estructural (accesión de Genbank MT586027).
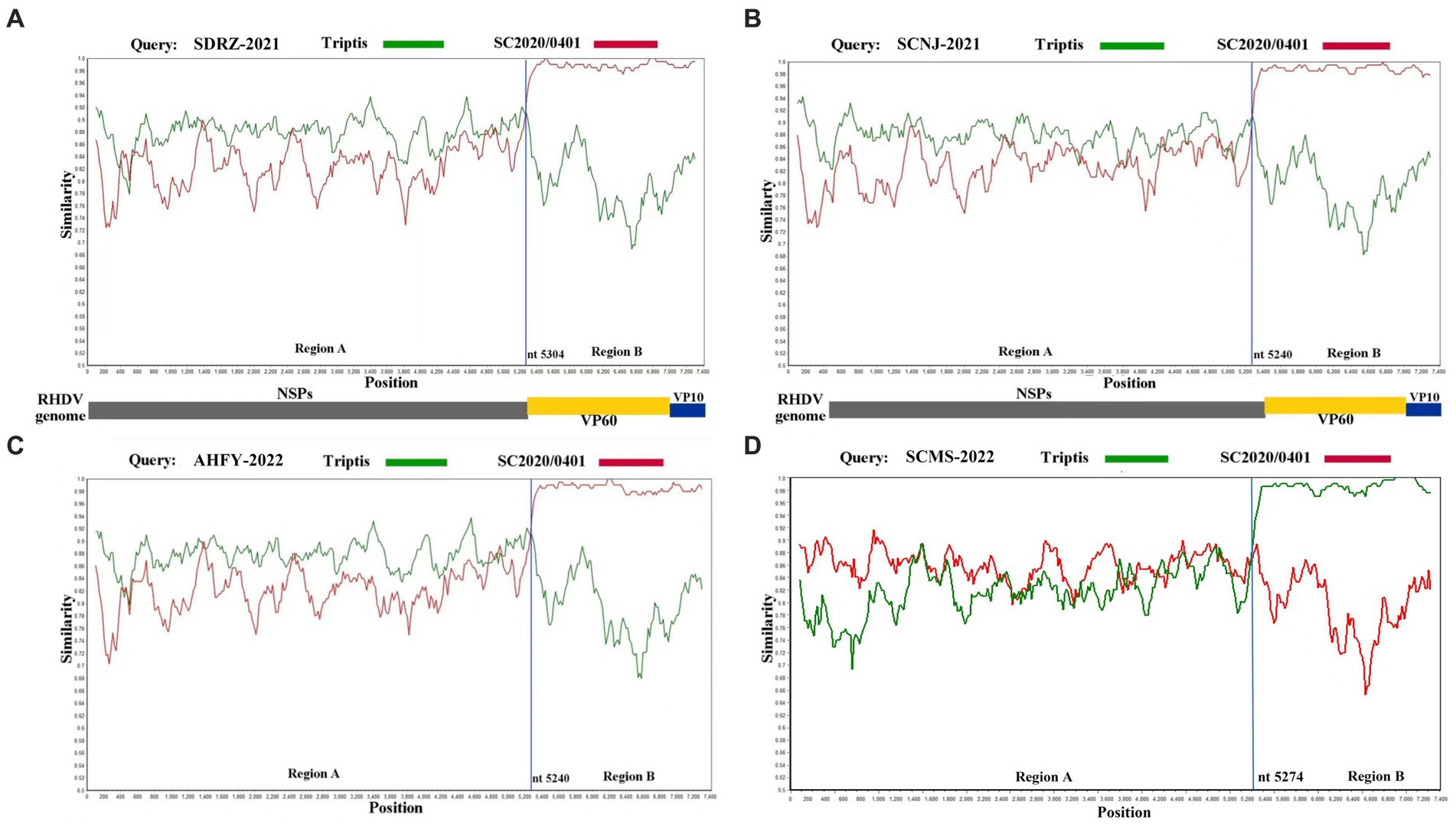 Figura 3. Análisis de recombinación de las cepas SDRZ-2021 (A), SCNJ-2021 (B), AHFY-2022 (C) y SCMS-2022 (D). El análisis se realizó utilizando una ventana deslizante de ventana de 200 pb y un paso de 20 pb. El eje y indica la similitud porcentual entre la secuencia de consulta y las secuencias de referencia. (A) Comparaciones de similitud de escala genómica de SDRZ-2021 (consulta) con Triptis (verde) y SC2020/0401 (rojo); (B) comparaciones de similitud de escala genómica de SCNJ-2021 (consulta) con Triptis (verde) y SC2020/0401 (rojo); (C) comparaciones de similitud de escala genómica de AHFY-2022 (consulta) con Triptis (verde) y SC2020/0401 (rojo); (D) comparaciones de similitud de escala genómica de SCMS-2022 (consulta) con Triptis (verde) y SC2020/0401 (rojo). Los puntos de ruptura de recombinación están marcados en la parte inferior con sitios de nucleótidos y estructura del genoma viral referenciada a SC2020/0401.
Figura 3. Análisis de recombinación de las cepas SDRZ-2021 (A), SCNJ-2021 (B), AHFY-2022 (C) y SCMS-2022 (D). El análisis se realizó utilizando una ventana deslizante de ventana de 200 pb y un paso de 20 pb. El eje y indica la similitud porcentual entre la secuencia de consulta y las secuencias de referencia. (A) Comparaciones de similitud de escala genómica de SDRZ-2021 (consulta) con Triptis (verde) y SC2020/0401 (rojo); (B) comparaciones de similitud de escala genómica de SCNJ-2021 (consulta) con Triptis (verde) y SC2020/0401 (rojo); (C) comparaciones de similitud de escala genómica de AHFY-2022 (consulta) con Triptis (verde) y SC2020/0401 (rojo); (D) comparaciones de similitud de escala genómica de SCMS-2022 (consulta) con Triptis (verde) y SC2020/0401 (rojo). Los puntos de ruptura de recombinación están marcados en la parte inferior con sitios de nucleótidos y estructura del genoma viral referenciada a SC2020/0401.
3.5. Protección cruzada entre las variantes GI.2 y GI.1aP-GI.2
Para demostrar si la vacuna preparada a partir de la cepa prototipo GI.2 confería protección frente a la variante GI.2 recombinante, se realizó el estudio de provocación. Como era de esperar, todos los conejos juveniles y adultos no vacunados murieron por el desafío con la variante GI.2 o GI.1aP-GI.2 de 24 h a 96 h después de la infección. Sin embargo, la vacunación con GI.2 (SCMS-2020) proporcionó una protección completa contra la infección GI.2 de los padres y una protección cruzada incompleta contra la infección GI.1aP-GI.2 (SCNJ-2021). La protección fue ligeramente menor en conejos jóvenes vacunados que en conejos adultos para la variante G1.1aP-GI.2 (SCNJ-2021) (Tabla 1), y todos los conejos sucumbieron a la infección por virus experimentaron un curso de enfermedad subagudo o agudo. El tiempo de supervivencia para los conejos desafiados con GI.2 (SCMS-2020) o GI.1aP-GI.2 (SCNJ-2021) tanto en grupos no vacunados como vacunados no fue significativamente diferente (Tabla suplementaria S5).
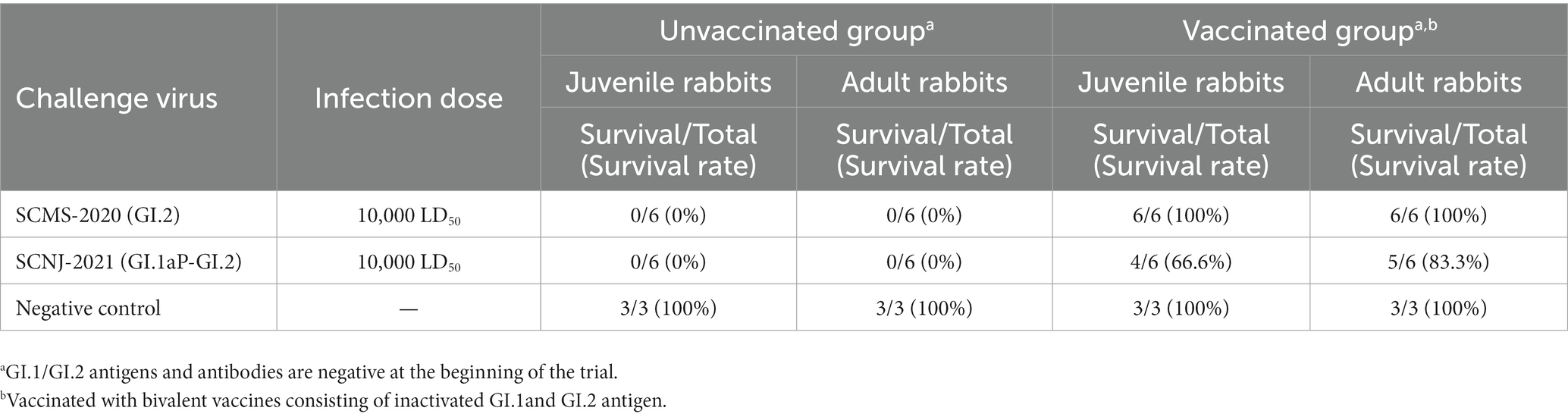 Tabla 1. Resultados del desafío utilizando prototipos GI.2 y GI.1aP-GI.2.
Tabla 1. Resultados del desafío utilizando prototipos GI.2 y GI.1aP-GI.2.
4. Discusión
4.1. La secuenciación de nucleótidos proporciona información para las nuevas variantes GI.1aP-GI.2 emergentes
Desde el primer aviso de cepas GI.2 en 2010 (Le Gall-Reculé et al., 2011), los eventos de recombinación en curso entre GI.2 y otros genotipos de lagovirus generaron varias variantes que surgieron como cepas prevalentes con amplia distribución en el mundo. Durante mucho tiempo, GI.1a había sido una vez la variante predominante en China hasta el brote de GI.2 en 2020. Cuando investigamos la presencia de GI.1 y GI.2 dentro del alcance del monitoreo rutinario de la enfermedad debido a los síntomas típicos y los hallazgos de necropsia en conejos muertos, se identificaron nuevas variantes GI.2 de muestras hepáticas sospechosas y se realizaron más investigaciones para determinar la diversidad genética de los genotipos GI.2. La unión RdRp-VP60 se considera un punto caliente de recombinación robusto, por lo tanto, la secuenciación de nucleótidos para esta región es un enfoque de detección rápida para el análisis de recombinación. La frecuencia de recombinación altamente intergenotípica (40,9%) de GI.1a y GI.2 indica que es probable que estas nuevas variantes sean las cepas predominantes en los años siguientes. Aunque este estudio puede subestimar la incidencia actual de estas variantes predominantes en la industria del conejo en China debido a la falta de datos de presentación. Los resultados respaldan firmemente la rápida aparición de las nuevas variantes epidémicas (GI.1aP-GI.2) desde el brote de GI.2 en China. Hasta donde sabemos, este patrón de recombinación nunca se ha descrito antes.
4.2. Recombinación genética inferida adaptación evolutiva de las variantes GI.1aP-GI.2
Los genes que codifican NSP de los lagovirus determinan el potencial de replicación del virus y la evasión inmune (Urakova et al., 2015; Zhu et al., 2022), que es un proceso complejo que promueve la evolución del virus con variación frecuente de nucleótidos (Silvério et al., 2018; Mahar et al., 2021). En este estudio, el análisis de recombinación revela que las variantes GI.1aP-GI.2 se ramifican en un solo clado, que posee orígenes moleculares similares de los GI.1a y GI.2 parentales, lo que indica que este nuevo evento de recombinación puede ocurrir en las cepas de RHDV de China después del brote de GI.2 en 2020 (Chen et al., 2022). Además, el alto contenido de nucleótidos dentro de estas cuatro variantes también demuestra que estas nuevas cepas recombinantes tienen una estrecha relación geográfica. Mientras tanto, la posición de los puntos de ruptura de recombinación en estas cuatro variantes es flexible, lo que da forma a la diversidad intragenotípica del patrón de recombinación GI.2 bajo la selección evolutiva. Sin embargo, el mecanismo de la recombinación entre GI.1a y GI.2 no está completamente dilucidado.
Se ha informado ampliamente que la exposición a prototipos de infección GI.1 y GI.2 en conejos podría promover una recombinación robusta entre estos dos genotipos en un contexto temporal y espacial a gran escala (Silvério et al., 2018; Abrantes y otros, 2020a; Al-Ebshahy et al., 2022). También se confirmaron resultados similares en conejos y liebres coinfectados con RHDV2 (GI.2) y el virus del síndrome de la liebre parda europea (EBHSV GII.1) (Le Gall-Reculé et al., 2017). Por lo tanto, la recombinación intergenotípica entre GI.1a y GI.2 puede indicar una nueva exaptación de contrapartes GI.2 en respuesta a la interacción coevolutiva entre el huésped y el virus (Schwensow et al., 2014). Este patrón de recombinación predominante en nuestros hallazgos revela que las variantes GI.1aP-GI.2 posiblemente adaptan ciertas estrategias de expansión de la población, que pueden atribuirse a la velocidad y fidelidad de RdRp, para obtener su ventaja evolutiva y persistencia entre las variantes GI.2 (Mahar et al., 2021). Además, la presión de selección posiblemente mediada por anticuerpos para estos dos virus también puede favorecer las variantes GI.1aP-GI.2 para reemplazar la GI.2 parental y adquirir una adaptabilidad de fortalecimiento (Hall et al., 2021; O’Connor et al., 2022; Patel et al., 2022). Sin embargo, la falta de evidencia directa de que la inmunidad del huésped contribuya a la recombinación GI.1aP-GI.2 justifica una investigación adicional.
4.3. Una nueva recombinación podría contribuir a alterar la virulencia de las variantes GI.1aP-GI.2
El impacto de la recombinación genética en la virulencia de las variantes GI.2 se ha investigado en estudios previos (Calvete et al., 2018; Müller et al., 2021). Se confirmaron las ocurrencias de protección cruzada de amplio espectro entre las variantes GI.1bP-GI.2, GI.4eP-GI.2 y GI.4cP-GI.2, independientemente de los patrones de recombinación y las dosis de desafío (Calvete et al., 2018; Müller y otros, 2021; O’Connor et al., 2022). Sin embargo, en nuestro estudio, la vacunación con GI.2 parental confiere una protección cruzada incompleta contra el desafío por variantes GI.1aP-GI.2 en conejos vacunados. Esto sugiere que la nueva variante puede escapar de la inmunidad adaptativa del huésped producida por GI.2 parental y evolucionar para desequilibrar la interacción huésped-virus (Mahar et al., 2016; Lopes et al., 2018).
Aunque no hay diferencia estadística entre el tiempo de supervivencia de los conejos en cada grupo, el aumento de la mortalidad y la menor LD50 valor para la variante GI.1aP-GI.2 todavía apoya nuestra hipótesis de que los mecanismos de recombinación distintivos pueden favorecer la variante GI.1aP-GI.2 para adquirir una patogenicidad moderadamente mejorada. Aunque surgió una gran cantidad de variación de nucleótidos y aminoácidos a lo largo del genoma de los recombinantes, no se confirmó claramente que los sitios de sustitución consistentes dentro de los genes no estructurales o estructurales estuvieran asociados con la alteración de la virulencia de las variantes GI.2 recombinantes (datos no mostrados), lo que también justifica un mayor estudio biológico e investigación bioinformática para dilucidar estas variaciones genéticas. De acuerdo con el manejo de biocontrol de lagomorfos silvestres en varios países, la exposición temprana al RCV no patógeno o GI.1 patógeno confirió protección parcial al desafío GI.2 (Patel et al., 2022; Taggart et al., 2022). Sin embargo, la rápida aparición de variantes recombinantes entre las cepas de cocirculación hizo que el programa de biocontrol de Lagomorphs silvestres fuera menos exitoso que antes, lo que también implica que la recombinación genética indeseable puede desactivar la protección inducida por la vacuna conferida por las cepas parentales de RHDV. Como es bien sabido, la proteína VP60 se considera un antígeno importante y determinante de virulencia del IG.2 (Miao et al., 2019; Liu et al., 2022). Curiosamente, en un estudio de encuesta, la aparición de recombinantes basados en GI.2 VP60 tenía más probabilidades de ser cepas predominantes entre las variantes intergenotípicas circulantes (Mahar et al., 2021), lo que sugiere la ventaja competitiva de los recombinantes GI.2 sobre otros genotipos debido a las frecuentes substiciones de aminoácidos a lo largo de los NSP, pero no su proteína VP60. Además, la disparidad molecular entre GI.1aP-GI.2 y otros recombinantes GI.2 también implica que los NSP no solo pueden ser relevantes para la aptitud del virus (Mahar et al., 2021), sino que también pueden estar asociados con la alteración de la virulencia hasta cierto punto.
Estos hallazgos respaldan la importancia de la variabilidad genética para la rápida propagación de las cepas GI.1aP-GI.2 en los conejos en China bajo presión ambiental y también implican el potencial de estas variantes para manipular la inmunidad del huésped.
5. Conclusión
Hasta donde sabemos, el nuevo GI.1aP-GI.2 recombinante se identificó por primera vez en conejos domésticos en China, lo que amplió el conocimiento sobre la filodinámica y la diversidad genómica de los genotipos GI.2. La rápida evolución molecular y la variada patogenicidad de estos virus recombinantes ponen de relieve la necesidad urgente de vigilancia epidemiológica y de prevención futura de las variantes GI.2 desatendidas.
Declaración de disponibilidad de datos
Los conjuntos de datos presentados en este estudio se pueden encontrar en repositorios en línea. Los nombres del repositorio / repositorios y el número (s) de acceso se pueden encontrar en el artículo / material complementario.
Declaración ética
El estudio en animales fue revisado y aprobado por el Comité de Ética del Centro Experimental Animal del Grupo Biológico Huapai (número de permiso: 2022HPAES018).
Contribuciones del autor
Diseño del estudio JH y YL. Implemente el experimento DD, YL, LS, CY y HZ. Análisis de datos LZ, JY y JH y revisión de manuscritos. Redacción de manuscritos YL y JH. Todos los autores han aprobado el manuscrito.
Financiación
Esta investigación cuenta con el apoyo de los Fondos de Inicio de Investigación de la Universidad del Suroeste de Minzu (Subvención No. RQD2021098), la Fundación de Ciencias Naturales de la Provincia de Sichuan (Subvención No. 2022NSFSC0081) y los Proyectos de Investigación Básica de los Institutos de Investigación Científica de Bienestar Público (Subvención No. SASA202302).
Reconocimientos
Los autores reconocen a Huapai Bio-engineering Group Co., Ltd (Chengdu, China) por proporcionar los conejos para este estudio, y reconocen a Zhang, R.H. (Facultad de Ciencia Animal y Medicina Veterinaria, Southwest Minzu University, Chengdu City, China.) por su asesoramiento lingüístico.
Conflicto de intereses
DD fue empleado por Huapai Bio-engineering Group Co., Ltd.
Los autores restantes declaran que la investigación se realizó en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un posible conflicto de intereses.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o reclamo que pueda ser hecho por su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1188380/full#supplementary-material
Referencias
Abrantes, J., Droillard, C., Lopes, A. M., Lemaitre, E., Lucas, P., Blanchard, Y., et al. (2020a). Recombinación ante la aparición del virus patógeno de la enfermedad hemorrágica del conejo Lagovirus europaeus/GI.2. 10:14502. DOI: 10.1038/S41598-020-71303-4
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Abrantes, J., Lopes, A. M., Lemaitre, E., Ahola, H., Banihashem, F., Clément Droillard, C., et al. (2020b). El análisis retrospectivo muestra que la mayoría de las cepas RHDV GI.1 circulantes desde finales de la década de 1990 en Francia y Suecia eran cepas GI.3P-GI.1d recombinantes. Genes (Basilea) 11:910. doi: 10.3390/genes11080910
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Aguayo-Adán, J. A., Rouco, C., Delibes-Mateos, M. y Santoro, S. (2022). Falta de pruebas de diferencias en la propagación de los virus clásicos (Lagovirus europaeus/GI.1) y nuevos (Lagovirus europaeus/GI.2) de la enfermedad hemorrágica del conejo en Europa y el norte de África. Rec. 190:E1067. DOI: 10.1002/VTR.1067
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Al-Ebshahy, E., Abas, O. y Abo-ElKhair, M. (2022). Cocirculación de los genotipos GI.1 y GI.2 del virus de la enfermedad hemorrágica del conejo en Egipto. Virus Dis. 33, 422–428. DOI: 10.1007/S13337-022-00791-X
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Calvete, C., Mendoza, M., Alcaraz, A., Sarto, M. P., Jiménez-de-Bagüéss, M. P., Calvo, A. J., et al. (2018). Enfermedad hemorrágica del conejo: protección cruzada y patogenicidad comparativa de los lagovirus GI.2/RHDV2/b y GI.1b/RHDV en un ensayo de provocación. Veterinario Microbiol. 219, 87–95. doi: 10.1016/j.vetmic.2018.04.018
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Chehida, F. B., Lopes, A. M., Côrte-Real, J. V., Sghaier, S., Aouini, R., Messadi, L., et al. (2021). Introducciones múltiples del virus de la enfermedad hemorrágica del conejo Lagovirus europaeus/GI.2 en África. Biología 10:883. doi: 10.3390/biología10090883
Chen, W., Tu, T., Luo, Y., Yang, Z. X., Yao, X. P., Wu, X. L., et al. (2022). Detection of a new emerging strain of rabbit haemorrhagic disease virus 2 (GI.2) in China. J. Vet. Res. 66, 289–295. doi: 10.2478/jvetres-2022-0048
Dalton, K. P., Nicieza, I., Balseiro, A., Muguerza, M. A., Rosell, J. M., Casais, R., et al. (2012). Variant rabbit hemorrhagic disease virus in young rabbits. Spain. Emerg. Infect. Dis. 18:2009. doi: 10.3201/eid1812.120341
Hall, R. B., King, T. G., O’Connor, T. W., Read, A. J., Vrankovic, S., Piper, M., et al. (2021). Passive immunisation against RHDV2 induces protection against disease but not infection. Vaccines 9:1197. doi: 10.3390/vaccines9101197
Hu, B., Fan, Z. Y., Wang, F., Song, Y. H., Wei, H. J., Liu, X., et al. (2016). A new variant of rabbit hemorrhagic disease virus G2-like strain isolated in China. Virus Res. 215, 20–24. doi: 10.1016/j.virusres.2016.01.018
Hu, Z. J., Tian, X. J., Zhai, Y. J., Xu, W., Zheng, D., and Sun, F. (2010). Cryo-electron microscopy reconstructions of two types of wild rabbit hemorrhagic disease viruses characterized the structural features of Lagovirus. Protein Cell 1, 48–58. doi: 10.1007/s13238-010-0007-0
Hu, B., Wang, F., Fan, Z. Y., Song, Y. H., Abrantes, J., Zuo, Y. Y., et al. (2017). Recombination between G2 and G6 strains of rabbit hemorrhagic disease virus (RHDV) in China. Arch. Virol. 162, 269–272. doi: 10.1007/s00705-016-3082-6
Hu, B., Wei, H. J., Fan, Z. Y., Song, Y. H., Chen, M. M., Qiu, R. L., et al. (2021). Emergence of rabbit haemorrhagic disease virus 2 in China in 2020. Vet. Med. Sci. 7, 236–239. doi: 10.1002/vms3.332
Le Gall-Reculé, G., Lemaitre, E., Bertagnoli, S., Hubert, C., Top, S., Decors, A., et al. (2017). Large-scale lagovirus disease outbreaks in European brown hares (Lepus europaeus) in France caused by RHDV2 strains spatially shared with rabbits (Oryctolagus cuniculus). Vet. Res. 48:70. doi: 10.1186/s13567-017-0473-y
Le Gall-Reculé, G., Zwingelstein, F., Boucher, S., Le Normand, B., Plassiart, G., Portejoie, Y., et al. (2011). Detection of a new variant of rabbit haemorrhagic disease virus in France. Vet. Rec. 168, 137–138. doi: 10.1136/vr.d697
Le Pendu, J., Abrantes, J., Bertagnoli, S., Guitton, J. S., Le Gall-Reculé, G., Lopes, A. M., et al. (2017). Proposal for a unified classification system and nomenclature of lagoviruses. J. Gen. Virol. 98, 1658–1666. doi: 10.1099/jgv.0.000840
Liu, C. J., Lin, M., Hu, H. Y., Liu, X. L., Bian, Y. C., and Deng, S. Z. (2022). Rabbit hemorrhagic disease virus VP60 protein expressed in recombinant swinepox virus self-assembles into virus-like particles with strong immunogenicity in rabbits. Front. Microbiol. 13:960374. doi: 10.3389/ fmicb. 2022. 960 374
Liu, S. J., Xue, H. P., Pu, B. Q., and Qian, N. H. (1984). A new viral disease in rabbit (in Chinese). Anim. Husb. Vet. Med. 16, 253–255.
Lopes, A. M., Breiman, A., Lora, M., Moullac-Vaidye, B. L., Galanina, O., and Nyström, K. (2018). Host-specific glycans are correlated with susceptibility to infection by Lagoviruses, but not with their virulence. J. Virol. 92, e01759–e01717. doi: 10.1128/JVI.01759-17
Lopes, A. M., Silvério, D., Magalhães, M. J., Areal, H., Alves, P. C., Esteves, P. J., et al. (2017). Characterization of old RHDV strains by complete genome sequencing identifies a novel genetic group. Sci. Rep. 7:13599. doi: 10.1038/s41598-017-13902-2
Mahar, J. E., Hall, R. N., Peacock, D., Kovaliski, J., Piper, M., Mourant, R., et al. (2018). Rabbit hemorrhagic disease virus 2 (RHDV2; GI.2). Is replacing endemic strains of RHDV in the Australian landscape within 18 months of its arrival. J. Virol. 92, e01374–e01317. doi: 10.1128/JVI.01374-17
Mahar, J. E., Jencke, M., Huang, N., Smertina, E., Holmes, E. C., Strive, T., et al. (2021). Frequent intergenotypic recombination between the non-structural and structural genes is a major driver of epidemiological fitness in caliciviruses. Virus. Evol. 7:veab080. doi: 10.1093/ve/veab080
Mahar, J. E., Nicholson, L., Eden, J. S., Duchêne, S., Kerr, P. J., Duckworth, J., et al. (2016). Los calicivirus benignos de conejo exhiben dinámicas evolutivas similares a las de sus parientes virulentos. J. Virol. 90, 9317–9329. doi: 10.1128/JVI.01212-16
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Miao, Q., Qi, R., Veldkamp, L., Ijzer, J., Kik, M. L., Zhu, J., et al. (2019). Inmunogenicidad en conejos de partículas similares a virus de un virus contemporáneo de la enfermedad hemorrágica del conejo tipo 2 (GI.2/RHDV2/b) aislado en los Países Bajos. Virus 11:553. DOI: 10.3390/v11060553
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Mizoguchi, T., Ken-ichi, I., y Sakurai, M. (2003). Hemaglutinación y comparación antigénica del virus de la enfermedad hemorrágica del conejo. J. Vet. Med. Sci. 65, 95–97. DOI: 10.1292/JVMS.65.95
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Müller, C., Hrynkiewicz, R., Bębnowska, D., Maldonado, J., Massimiliano Baratelli, M., Köllner, B., et al. (2021). Inmunidad frente a Lagovirus europaeus y el impacto de los estudios inmunológicos en la vacunación. Vacunas 9:255. doi: 10.3390/vaccines9030255
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Neimanis, A. S., Ahola, H., Zohari, S., Pettersson, U. L., Bröjer, C., Capucci, L., et al. (2018b). Llegada del virus de la enfermedad hemorrágica del conejo 2 al norte de Europa: aparición y brotes en conejos salvajes y domésticos (Oryctolagus cuniculus) en Suecia. Transbound. Emerg. Dis. 65, 213–220. doi: 10.1111/tbed.12650
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Neimanis, A. S., Pettersson, U. L., Huang, N., Gavier-Widén, D. y Strive, T. (2018a). Elucidación de la patología y distribución tisular de Lagovirus europaeus GI.2/RHDV2 (virus de la enfermedad hemorrágica de conejo 2) en conejos jóvenes y adultos (Oryctolagus cuniculus). Vet. 49:46. doi: 10.1186/s13567-018-0540-z
Resumen de PubMed | Texto completo de CrossRef | Google Académico
O’Connor, T. F., Read, A. J., Hall, R. N., Strive, T. y Kirkland, P. D. (2022). Protección inmunológica cruzada entre diferentes virus de enfermedades hemorrágicas de conejos: implicaciones para el biocontrol de conejos y el desarrollo de vacunas. Vacunas 10:666. doi: 10.3390/vaccines10050666
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Pacioni, C., Hall, R. N., Strive, T., Ramsey, D. S. L., Gill, M. S. y Vaughan, T. G. (2022). Epidemiología comparativa de cepas del virus de la enfermedad hemorrágica del conejo a partir de datos de secuencias virales. Virus 15:21. DOI: 10.3390/v15010021
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Patel, K. P., Strive, T., Hall, R. N., Mutze, G., Page, B., Korcz, M., et al. (2022). Tasas de protección cruzada, infección y letalidad en conejos europeos salvajes desafiados experimentalmente con diferentes virus de enfermedades hemorrágicas de conejo. Transbound. Emerg. Dis. 69, e1959–e1971. doi: 10.1111/tbed.14530
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Qi, R. B., Meng, C. C., Zhu, J., Li, H., Miao, Q. H., Tang, J. Y., et al. (2022). El brote del virus hemorrágico del conejo tipo 2 en el interior de China puede estar relacionado con el semen importado. Virol. Pecado. 37, 623–626. doi: 10.1016/j.virs.2022.04.003
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Schwensow, N. I., Cooke, B., Kovaliski, J., Sinclair, R., Peacock, D., Fickel, J., et al. (2014). Enfermedad hemorrágica del conejo: persistencia y adaptación del virus en Australia. Evol. Appl. 7, 1056–1067. doi: 10.1111/eva.12195
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Silvério, D., Lopes, A. M., Melo-Ferreira, J., Magalhães, M. J., Monterroso, P., Serronha, A., et al. (2018). Información sobre la evolución de la nueva variante del virus de la enfermedad hemorrágica del conejo (GI.2) y la identificación de nuevas cepas recombinantes. Transbound. Emerg. Dis. 65, 983–992. doi: 10.1111/tbed.12830
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Smertina, E., Hall, R. N., Urakova, N., Strive, T. y Frese, M. (2021). Proteínas no estructurales de Calicivirus: funciones potenciales en la replicación y manipulación de la célula huésped. Frente. Microbiol. 12:2021. doi: 10.3389/fmicb.2021.712710.eCollection
Song, Y. H., Fan, Z. Y., Zuo, Y. Y., Wei, H. J., Hu, B., Chen, M. M., et al. (2017). La unión de partículas similares al virus de la enfermedad hemorrágica del conejo a los antígenos del grupo histosanguíneo huésped es bloqueada por antisueros de conejos vacunados experimentalmente. Arq. Virol. 162, 3425–3430. DOI: 10.1007/S00705-017-3509-8
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Taggart, P. L., Hall, R. N., Cox, T. E., Kovaliski, J., McLeod, S. R. y Strive, T. (2022). Los cambios en la dinámica de transmisión del virus tras la aparición de RHDV2 arrojan luz sobre su ventaja competitiva sobre las variantes que circulaban anteriormente. Transbound. Emerg. Dis. 69, 1118–1130. doi: 10.1111/tbed.14071
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Teifke, J. P., Reimann, I. y Schirrmeier, H. (2002). Necrosis hepática subaguda después de la infección experimental con el virus de la enfermedad hemorrágica del conejo (RHDV). J. Comp. Pathol. 126, 231–234. DOI: 10.1053/JCPA.2001.0534
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Urakova, N., Frese, M., Hall, R. N., Liu, J., Matthaei, M. y Strive, T. (2015). Expresión y caracterización parcial de proteínas no estructurales del virus de la enfermedad hemorrágica del conejo. Virología 484, 69–79. doi: 10.1016/j.virol.2015.05.004
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Wang, X. L., Hao, H. F., Qiu, L., Dang, R. Y., Du, E. Q., Zhang, S. X., et al. (2012). Análisis filogenético del virus de la enfermedad hemorrágica del conejo en China y la variación antigénica de nuevas cepas. Arq. Virol. 157, 1523–1530. DOI: 10.1007/S00705-012-1340-9
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zhou, J., Ma, Y. J., Wang, M., Zhang, Y., Chen, B., Chen, D. S., et al. (2022). Establecimiento de un TaqMan RT-PCR dúplex para la detección diferencial de RHDV GI.1 y GI.2. J Virol. Métodos 304:114526. doi: 10.1016/j.jviromet.2022.114526
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zhu, J., Miao, Q. H., Guo, H. Y., Tang, A. X., Dong, D. D., Tang, J. Y., et al. (2022). La nucleolina interactúa con la enfermedad hemorrágica del conejo replica el virus RdRp, proteínas no estructurales p16 y p23, desempeñando un papel en la replicación del virus. Virol. Pecado. 37, 48–59. doi: 10.1016/j.virs.2022.01.004
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Cita: Li Y, Du D, Zhou L, Su L, You C, Zhang H, Yu J, Xiao L y Huang J (2023) Primer informe de GI.1aP-GI.2 recombinantes del virus de la enfermedad hemorrágica del conejo en conejos domésticos en China. Frente. Microbiol. 14:1188380. doi: 10.3389/fmicb.2023.1188380
Editado por:
Shailendra Saxena, Universidad Médica del Rey Jorge, India
Revisado por:
Ángel L. Álvarez, Universidad de Oviedo, España
Margarida Duarte, Instituto Nacional de Investigaciones Agrarias y Veterinarias (INIAV), Portugal
Lauro Velázquez-Salinas, Servicio de Investigación Agrícola (USDA), Estados Unidos
Derechos de autor © 2023 Li, Du, Zhou, Su, You, Zhang, Yu, Xiao y Huang. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia de Atribución Creative Commons (CC BY).
*Correspondencia: Jian Huang, hjvet03@sina.cn
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o reclamo que pueda ser hecho por su fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía