
Uso de la secuenciación del genoma completo para desentrañar la diversidad genética de un Spoligotipo de Prevalente Mycobacterium bovis en un escenario multihost en España
Uso de la secuenciación del genoma completo para desentrañar la diversidad genética de un Spoligotipo de Prevalente Mycobacterium bovis en un escenario multihost en España
 Javier Bezos1,2, Lucía de Juan1,2, José Luis Saez4, Beatriz Romero1,2,
Javier Bezos1,2, Lucía de Juan1,2, José Luis Saez4, Beatriz Romero1,2, Julio Álvarez1,2 y en nombre de la Red Española de Vigilancia y Monitoreo de la Tuberculosis Animal
Julio Álvarez1,2 y en nombre de la Red Española de Vigilancia y Monitoreo de la Tuberculosis Animal- 1Centro de Vigilancia Sanitaria VISAVET, Universidad Complutense de Madrid, Madrid, España
- 2Departamento de Sanidad Animal, Facultad de Veterinaria, Universidad Complutense de Madrid, Madrid, España
- 3Laboramentos Nacionales de Servicios Veterinarios, Servicio de Inspección de Sanidad Animal y Vegetal, Departamento de Agricultura, Ames, IA, Estados Unidos
- 4Subdirección General de Sanidad e Higiene Animal y Trazabilidad, Dirección General de Sanidad de la Producción Agraria, Ministerio de Agricultura, Pesca y Alimentación, Madrid, España
A pesar de los esfuerzos invertidos en la erradicación de la tuberculosis bovina en España, la prevalencia del rehado se ha mantenido constante en el país durante los últimos 15 años (~1,5-1,9%) debido a una combinación de factores epidemiológicos que perjudican el control de la enfermedad, incluida la transmisión entre especies. Aquí, nuestro objetivo era investigar la diversidad molecular de los aislados de Mycobacterium bovis pertenecientes al altamente prevalente tipo SB0339 spoligotype en la interfaz ganado-wild en diferentes regiones de España utilizando la secuenciación del genoma completo (WGS). Los datos genómicos de 136 aislados de M. bovis recuperados de diferentes especies animales (tiga, jabalí, ciervos de barbecho y ciervo rojo) y ubicaciones entre 2005 y 2018 se analizaron para investigar la transmisión entre y dentro de las especies, así como dentro de los rebaños. Todos los aislados secuenciados diferían en 49-88 polimorfismos de un solo nucleótido de su antepasado común más reciente. La heterogeneidad genética era geográfica en lugar de específica de la especie huésped, ya que los aislados recuperados tanto del ganado como de la vida silvestre de una región determinada estaban más estrechamente relacionados en comparación con los aislados de la misma especie, pero geográficamente distantes. De hecho, se encontró una fuerte asociación entre las distancias geográficas y genéticas que separan los pares de aislados de M. bovis, con un efecto significativamente más fuerte cuando se comparan los aislados de ganado con los aislados de vida silvestre o de vida silvestre en España. Los mismos resultados se obtuvieron en Madrid, la región con el mayor número de aislados secuenciados, pero no se observaron diferencias dependiendo del huésped. La diversidad genética dentro del rebaño fue limitada a pesar del tiempo considerable transcurrido entre los aislamientos. La detección de cepas estrechamente relacionadas en diferentes huéspedes demuestra la compleja dinámica de transmisión entre huéspedes presente en las zonas endémicas de España. En conclusión, el WGS es una herramienta valiosa para rastrear la infección por bTB a alta resolución y puede contribuir a lograr su erradicación en España.
Introducción
Mycobacterium bovis, un miembro del complejo Mycobacterium tuberculosis (MTBC) y el principal agente causante de la tuberculosis bovina (bTB), puede infectar una amplia gama de especies de mamíferos, incluidas las especies domésticas y silvestres, así como las humanas (Cousins, 2001; Aranaz et al., 2004; Perez-Lago et al., 2014). Si bien la enfermedad se ha erradicado con éxito en varios países utilizando estrategias de prueba y sacrificio y restricciones de movimiento (Reviriego Gordejo y Vermeersch, 2006; Panel de la EFSA sobre Salud y Bienestar Animal (AHAW), 2014; More et al., 2015), la prevalencia del rehado de bTB sigue siendo relativamente alta en varias regiones del mundo Los esfuerzos de erradicación se han visto constantemente obstaculizados por el largo período de latencia característico de la enfermedad (Goodchild y Clifton-Hadley, 2001), las limitaciones de las pruebas de diagnóstico disponibles, en particular para la detección de las primeras etapas de infección (de la Rua-Domenech et al., 2006), y la existencia de reservorios de vida silvestre (Garnett et Por esta razón, en lugares donde se sabe que los embalses de vida silvestre son importantes en la transmisión de bTB, el control de las enfermedades debe centrarse en un enfoque integrado en la interfaz de ganado-salvaje.
En España, a pesar de que la prevalencia del rebante ha disminuido significativamente desde la implementación inicial de los programas nacionales de erradicación en los años 80, la erradicación completa de la enfermedad sigue siendo esquiva (un 0,2 % de prevalencia del rebante en 2002 y el 1,6 % en 2020; Informe Final Técnico Tuberculosis Bovina Año, 2020). La tuberculosis bovina no se distribuye uniformemente por todo el país, ya que la prevalencia del rehado es mayor en las regiones central y meridional (hasta el 10,9 y el 6,4 % en Castilla-La Mancha y Andalucía, respectivamente en 2020), mientras que la enfermedad está prácticamente ausente en las partes del noroeste de España (un % de prevalencia del rehado en Galicia, Asturias y las Islas En las áreas endémicas de bTB, la distribución geográfica de las especies de ganado y vida silvestre que se sabe que están infectadas con M. bovis a menudo se superpone. Esto es particularmente relevante en el caso del ganado ampliamente gestionado, que puede compartir pastos y/o puntos de riego con especies de vida silvestre, lo que conduce a un riesgo potencial de transmisión interespecies de la infección. Además, en ciertos casos la infección residual puede persistir con el tiempo en un redero a pesar de la aplicación de programas de prueba y matanza debido a la presencia de animales infectados pero no detectados (Karolemeas et al., 2011; Guta et al., 2014b). Aunque el jabalí es el embalse de vida silvestre más importante de bTB en el centro y sur de España, los ciervos rojos y los ciervos de yoque también pueden servir como huéspedes potenciales (Gortazar et al., 2008; Naranjo et al., 2008).
El tipo de tipo de oligonucleótidos espaciador de repetición variable directa (spoligotipado DVR) y la unidad repetitiva intercalada micobacteral – tipografía de número variable de repetición en tándem (MIRU-VNTR) son las técnicas más utilizadas para caracterizar los aislados de M. bovis (Kamerbeek et al., La mayoría de los epoligotipos de M. bovis en España están circunscritos a regiones específicas, mientras que una pequeña proporción de los perfiles altamente prevalentes están muy extendidos en el país (es decir, SB0121, SB0134 o SB0339; Rodríguez-Campos et al., 2012). El mecanografía molecular de las cepas de M. bovis ha demostrado que tanto las especies de ganado como las especies de vida silvestre comparten perfiles similares de spoligotipo y VNTR, con agrupamiento espacial entre los huéspedes que sugiere una transmisión interespecífica a escala local (Aranaz et al., 2004). En estas circunstancias, solo se puede lograr un control efectivo de la bTB si también se tiene en cuenta la vida silvestre en los esfuerzos de erradicación. Además, la caracterización molecular puede ayudar a determinar si los animales recién detectados en rebaños previamente infectados pueden representar una reinfección o un fracaso en la eliminación de todo el ganado infectado en las pruebas de rebaño anteriores.
A pesar del uso generalizado de spoligotyping, esta herramienta por sí sola no puede reconstruir con precisión la filogenia de aislamientos de M. bovis o diferenciar aislamientos no relacionados epidemiológicamente en ciertos entornos debido a su resolución limitada y alta tasa de homoplasia (Comas et al., 2009). Los avances recientes en la secuenciación del genoma completo (WGS) permiten la realización de exámenes de detección de todo el genoma para estudiar poblaciones microbianas, lo que aumenta el poder de los estudios de epidemiología molecular. En este sentido, el uso de genotipado basado en polimorfismos de un solo nucleótido (SNP) puede proporcionar información valiosa sobre la patogenicidad y la evolución de las cepas de M. bovis, lo que permite la identificación de huéspedes o asociaciones espaciales, mejorar las investigaciones de brotes y la diferenciación de M. bovis. cepas en linajes y estructuras filogenéticas relacionadas (Joshi et al., 2012; Hauer et al., 2015; Ghebremariam et al., 2016; Trewby et al., 2016; Orloski et al., 2018; Price-Carter et al., 2018).
En el contexto de la erradicación y el control de la tuberculosis bovina, el seguimiento de la variabilidad de M. bovis dentro del hato es fundamental, ya que puede proporcionar información sobre posibles introducciones nuevas en eventos de recurrencia del hato. Además, comprender los patrones de transmisión de la tuberculosis bovina en el ganado bovino y entre el ganado bovino y las especies silvestres de diferentes áreas de España es crucial para evaluar el riesgo de infección para el ganado en una interfaz multihuésped, particularmente en áreas endémicas donde coexisten animales infectados y animales silvestres. Hasta la fecha, no se han realizado estudios multirregionales para caracterizar la diversidad genética entre aislados de M. bovis de ganado y fauna silvestre en España mediante WGS. Por estas razones, el objetivo de este estudio fue utilizar tecnologías WGS para evaluar el riesgo de transmisión de la tuberculosis bovina entre el ganado y la fauna silvestre utilizando uno de los espoligotipos más prevalentes en España, SB0339, como ejemplo de trabajo. Este espoligotipo se encuentra ampliamente distribuido por todo el país y está especialmente agrupado en la provincia de Madrid, con >48% del total de aislamientos SB0339 (extraído de mycoDB.es). Los objetivos específicos fueron (I) investigar la diversidad genómica entre los aislados de SB0339 M. bovis recuperados de ganado y fauna silvestre durante 2005-2018 de diferentes regiones de España en general, y en Madrid en particular; (II) reconstruir las relaciones filogenéticas entre los aislados; y (III) realizar un análisis comparativo de la diversidad genómica intraespecies e interespecies para comprender los procesos evolutivos subyacentes de M. bovis en la interfaz ganado-fauna silvestre en España.
Materiales y métodos
Aislar la selección y los métodos de laboratorio
Como parte del programa de erradicación de bTB, se recogen muestras de tejido de ganado o vida silvestre que se sospecha que están infectados con bTB debido a resultados positivos en las pruebas ante mortem (ganados) o la detección de lesiones que aparecen granulomatos en la inspección post mortem (ganados y vida silvestre) y se presentan a los Laboratorios de Referencia regionales para el cultivo bacteri Una vez que se detecta el crecimiento bacteriano, el ADN se extrae de los cultivos y la presencia de miembros de MTBC se confirma mediante métodos moleculares convencionales, como la PCR o el espoligotipado DVR (Cousins et al., 1991; Kamerbeek et al., 1997). En este contexto, se consideró una selección de aislados de M. bovis recuperados de muestras de tejido de ganado y vida silvestre infectados naturalmente utilizando procedimientos de aislamiento estándar en el Centro de Vigilancia Sanitaria VISAVET para su inclusión en los análisis realizados aquí.
El SB0339 fue seleccionado para el estudio, ya que este patrón es el tercer tipo spoligo más frecuente en España [8,1% de todos los aislados, precedido solo por SB0121 (28,2%) y SB0134 (10,7%)], y el poligotipo aislado con más frecuencia en Madrid (Rodriguez-Campos et al., 2010, 2012). Además, este tipo de poligo se detecta con frecuencia tanto en el ganado como en la vida silvestre (es decir, ciervos rojos, ciervos en barbecho y jabalíes). Un total de 1.501 (44% del total de cepas SB0339 en España, mycoDB.es) Las muestras de SB0339 estaban disponibles para su selección en la colección de cepas del Centro de Vigilancia Sanitaria VISAVET y se dividieron en estratos basados en las especies animales de aislamiento, la ubicación (provincia) y el año de aislamiento. Posteriormente, se realizó un muestreo aleatorio estratificado, de modo que solo se consideraron para la selección subconjuntos de aislados SB0339 recuperados de provincias y años en los que había más de una especie huésped disponible. En consecuencia, 259 aislados del tipo SB0339 spoligotype recuperados del ganado y la vida silvestre entre 2005 y 2018 en 21 regiones de España fueron sometidos a recultura bacteriológica. Además, para evaluar el grado de heterogeneidad genética entre los aislados de M. bovis en rehados infectados por bTB, se seleccionaron entre 2 y 8 aislados de 15 rehadas. Diez de estas manadas habían sido infectadas crónicamente, definidas como rebanadas con aislamiento de M. bovis en más de 1 año.
Los aislamientos seleccionados identificados como M. bovis se centrifugaron a 2500 × g durante 10 min y, posteriormente, se lavaron dos veces con 5 ml de solución salina tamponada con fosfato (PBS, Gibco) y se centrifugaron. Se vertieron los sobrenadantes y los sedimentos se resuspendieron en 4 ml de PBS. Las muestras se inactivaron y el ADN micobacteriano se separó aún más de los otros componentes celulares utilizando un protocolo basado en disrupción de perlas y fenol/cloroformo/alcohol isoamílico (PCI, Sigma-Aldrich) como se describe en otro lugar.1 La calidad y la concentración del ADN se midieron utilizando una nanogota espectrofotómetro y un fluorómetro Qubit™ (Invitrogen).
Secuenciación de genoma completo
El ADN micobacteriano extraído se presentó al Laboratorio Nacional de Servicios Veterinarios (NVSL) en Ames, Iowa (Estados Unidos) para realizar WGS. Las bibliotecas se prepararon utilizando el kit de preparación Nextera XT y el ADN genómico total se secuenciaba en un instrumento MiSeq para producir lecturas de 2 × 250 bp (Illumina, San Diego, CA, Estados Unidos). Los archivos FASTQ generados se analizaron utilizando la tubería vSNP interna del Departamento de Agricultura de los Estados Unidos (USDA) NVSL, una variante de referencia de alta resolución que llama tubería.2 En resumen, las lecturas genómicos se mapearon con el genoma de referencia M. bovis AF2122/97 (número de acceso del Centro Nacional de Información Biotecnológica Se utilizó la definición de SNP para identificar diferentes grupos de aislados dentro de la canalización vSNP como se especifica en los archivos de dependencia vSNP.
Los polimorfismos de un solo nucleótido se llamaron usando FreeBayes, un detector de variantes basado en haplotipos, que genera archivos de formato de llamada de variantes (VCF; Garrison y Marth, 2012). Los resultados se filtraron utilizando una puntuación mínima de calidad a escala de doctorado (QUAL) de 150 y un recuento de alelo (AC) de 2. Los aislados que contenían llamadas heterociólogas/heterocigóticas en una posición de SNP (AC = 1 y presentes en <90% de las lecturas) se consideraron ambiguos como codificados por la Unión Internacional de Química Pura y Aplicada (IUAPC) y fueron inspeccionados visualmente. Aquellas posiciones de SNP que tenían una llamada variante en más del 90% de las lecturas se consideraron homocigóticas, mientras que las llamadas variantes identificadas en <90% de las lecturas de la muestra se consideraron heterocigotas y se eliminaron del análisis.
A continuación, se generó un resumen de las métricas de calidad para evaluar el rendimiento de la secuenciación de cada aislado. Esto incluyó la profundidad media de la cobertura, la longitud media de lectura, el porcentaje del genoma de referencia cubierto por las lecturas de cada aislado, el número de contigs que no se asignan a la referencia, el número de SNP con una puntuación de calidad (QUAL) de >300 con un AC de 2 (buenos SNP) y el código octal de tipo s El código octal se basó en los recuentos de cada secuencia espaciadora contra los archivos FASTQ sin procesar. Las lecturas identificadas como el complejo de M. tuberculosis se aislaron y las lecturas limpias se realizaron a través de vSNP.
Las tablas de SNP y los árboles filogenéticos se crearon después de eliminar todos los SNP no informativos (es decir, homogéneos/monórficos entre los aislados). Esos SNP identificados en el ∼10% del genoma compuesto por regiones repetitivas fueron excluidos utilizando archivos de enmascaramiento predeterminados en las dependencias de vSNP, ya que el mapeo en estas regiones es propenso a errores (Cole et al., 1998). Esto incluyó la familia de genes de prolina-glutamato (PE)/prolina-prolina-glutamato (PPE) altamente rica en GC y polimórfico (Cole et al., 1998). Además, se omitieron los SNP presentes en áreas con una acumulación anómala de variantes (normalmente ≥2 SNP en 10 bp debido a una mala alineación). Después de eliminar todas las posiciones variantes no informativas y potencialmente erróneas, los SNP informativos se validaron visualizando los archivos de alineación en el software Integrative Genomics Viewer (IGV) (Robinson et al., 2011).
Se construyeron árboles filogenéticos de máxima probabilidad con RAxML (Stamatakis, 2014) utilizando el archivo de alineación SNP que contiene los SNP polimórficos concatenados y validados. Se asumió que un modelo CAT reversible en tiempo general (GTR) para la tasa de sustitución de nucleótidos con una distribución Gamma tenía en cuenta la heterogeneidad entre sitios (Stamatakis, 2014). La precisión del árbol filogenético se confirmó utilizando la tabla SNP validada manualmente, y se realizó un filtrado adicional de SNP cuestionables de forma aislada por aislada cuando fuera apropiado.
El grado de relación genética entre los genomas de M. bovis se evaluó entre especies y dentro de las especies utilizando las distancias genéticas por pares entre los aislados. Los aislados se identificaron utilizando el año de aislamiento seguido del número de aislamiento (#), las iniciales de la provincia de origen, la especie animal y, si corresponde, el código del retablo o la herencia de la que se recuperó la muestra.
Análisis filogenéticos entre y dentro de las especificaciones
El análisis filogenético se realizó por primera vez en el número total de aislados disponibles para el estudio. Las distribuciones de las distancias de SNP por pares entre y dentro de los aislados de ganado y vida silvestre se compararon utilizando la prueba Kruskal-Wallis seguida de pruebas post-hoc con correcciones de Bonferroni para múltiples comparaciones. Se realizó una agrupación bayesiana jerárquica para determinar la estructura de la población utilizando BAPS (Cheng et al., 2013). Los aislados se consideraron genéticamente cercanos cuando sus secuencias estaban dentro de 0-3 SNP entre sí (Hatherell et al., 2016). La distancia geográfica en kilómetros entre pares de aislados se calculó utilizando los centróides de los municipios en los que se recuperaron los aislados, y la relación entre las distancias genéticas y geográficas se evaluó utilizando la prueba de correlación rho de Spearman.
Posteriormente, y para evitar un aumento artificial de la homogeneidad del ganado dentro de las especies, solo se seleccionó al azar un aislamiento por rebaño. La relación entre la distancia genética entre dos aislados y la distancia entre sus orígenes geográficos (provincia, municipio y/o estado) se exploró utilizando modelos de regresión lineal que también consideraban la especie de origen del huésped (es decir, si el par de aislados ambos se originaron del ganado, de la vida silvestre o de ambos). El modelo consideró los factores de riesgo seleccionados (distancia geográfica en km y especies huésped) junto con la interacción entre los dos. Finalmente, el mismo análisis se realizó solo en aislados de la región de Madrid, de los cuales se disponía del mayor número de aislados (ver resultados). Dado que solo un aislado por rebaño se seleccionó al azar en rebaños de ganado con múltiples aislados, el impacto de la selección en la evaluación de la asociación entre la distancia genética y el origen geográfico y la especie huésped se evaluó a través de un análisis de sensibilidad que consiste en repetir 10 veces los análisis con un aislado por rebaño muestreado intensivo seleccionado al azar.
Análisis filogenéticos dentro y entre el rebaño
Se seleccionaron aislados recuperados de diferentes rebaños de ganado para evaluar la variabilidad genética de los aislados recuperados de los mismos rebaños a lo largo del tiempo. Se recuperaron datos sobre el tiempo transcurrido entre los aislados y se disponía del número de años con aislamiento para cada retría infectada crónicamente. Se evaluaron las distancias genéticas dentro del rebaño (es decir, entre los aislados que se originan de una sola manada) teniendo en cuenta todos los rebaños y solo aquellos infectados crónicamente (con aislamientos de bTB recuperados en más de 1 año), junto con el número de cepas estrechamente relacionadas (≤3 SNP) presentes en los re Se calculó la correlación entre las distancias genéticas medias y el número de años diferentes de los que se recuperaron los aislados en cada retría.
Se realizaron comparaciones múltiples, gráficos y análisis de filogenia utilizando ggplot2 (Wickham, 2009), dplyr (Wickham et al., 2019), ape (Paradis et al., 2004), rgdal (Bivand et al., 2013), rhierbaps ( Tonkin-Hill et al., 2018) y paquetes PMCMRplus (Pohlert, 2020) para el software R (R Core Team, 2019).
Resultados
Análisis descriptivo
Se recuperaron ciento treinta y seis aislados de las 259 muestras seleccionadas de SB0339. De estos, el 72,8% (n = 99) se derivaron de la recolección de cepas VISAVET, mientras que el 27,2% restante fueron proporcionados por otros Laboratorios Regionales. Estas 136 cepas se obtuvieron durante el período 2005-2018 de ganado (72,8%, n = 99), jabalí (14,7%, n = 20), ciervos en barbecho (6,6%, n = 9) y ciervos rojos (5,9%, n = 8, Tabla 1). Se disponía de un número medio de 10 muestras por año (rango 2-4-25), con más aislados incluidos después de 2009, y los años 2010 y 2017 que representaron el mayor número de aislados (n = 17 y n = 25, respectivamente, Figura Suplementaria S1). Las áreas de las que se originaron las muestras utilizadas en el estudio incluyeron las regiones noroeste, noreste, central, oriental y sur del país (Tabla 1; Figura 1). La información de aislamiento individual y los metadatos asociados se detallan en la Tabla Suplementaria S1.La proporción más alta (48,5%, n = 66) de aislados se derivó de animales infectados por bTB ubicados en Madrid, seguido de Ciudad Real (6,6%, n = 9), Islas Baleares (a partir de ahora, la región de Mallorca, 5,9%, n = 8), Zaragoza (5,9%, n = 8) y Toledo (5,1%, n = 7). El resto de las provincias representaron los 38 aislamientos restantes (27,9%) (Tabla 1; Figura 1). La información sobre el municipio exacto del que se originaron los aislados estaba disponible en 121 aislados (82 %).
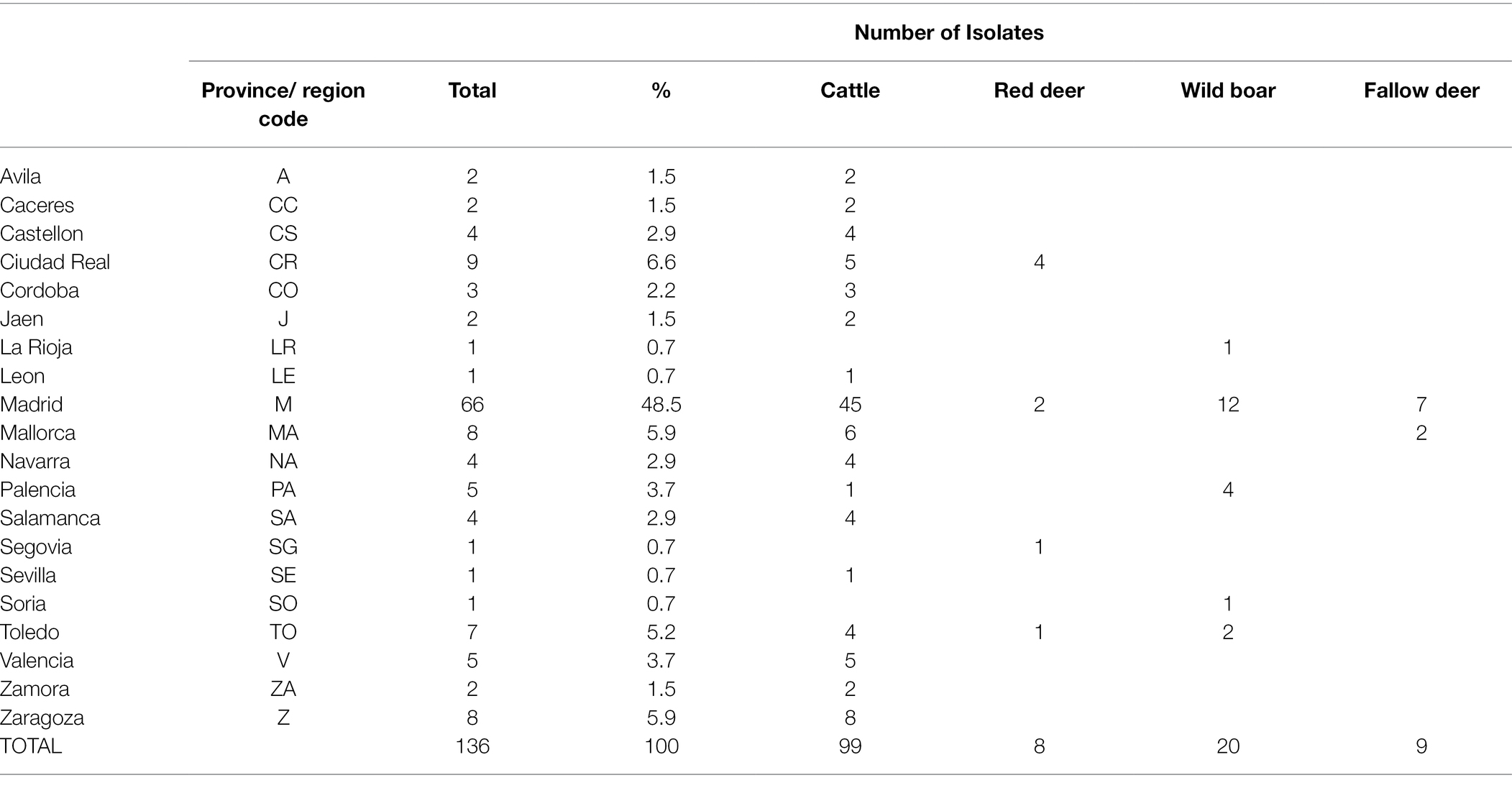 TABLA 1. Número de ganado, ciervos rojos, jabalíes y ciervos en barbecho secuenciados aislados por provincia y en general incluidos en el estudio.
TABLA 1. Número de ganado, ciervos rojos, jabalíes y ciervos en barbecho secuenciados aislados por provincia y en general incluidos en el estudio.
 FIGURA 1. Mapa del número de aislados de M. bovis SB0339 recuperados del ganado (A) y la vida silvestre (B) por provincia incluida en el estudio.
FIGURA 1. Mapa del número de aislados de M. bovis SB0339 recuperados del ganado (A) y la vida silvestre (B) por provincia incluida en el estudio.
Datos geómicos de Mycobacterium bovis
La profundidad media de la cobertura osciló entre 20x y 174x excepto 7 aislados, con un 85,3% (n = 116) de los aislados con valores ≥30x. La longitud media de lectura de los aislados osciló entre 186,5 y 239,4 (mediana = 229,7, IQR = 222.6-23,6). La cobertura media del genoma fue del 99,0% (IQR = 98,9-99),1). Los datos resumidos de alineación y los metadatos asociados con muestra de los 136 aislados sometidos a WGS se incluyen en la Tabla Suplementaria S1. No se detectó ninguna indicación de la presencia de aislados mixtos en la población secuenciada, ya que no se observaron muchas ocurrencias de posiciones mixtas de forma recurrente en todo el genoma en regiones con buenas puntuaciones de mapeo.
Análisis filogenético y estructura de agrupación
Se excluyeron unascientos once SNP, ya que se detectaron en regiones donde el mapeo era propenso a errores o estaban presentes en áreas con una acumulación anómala de variantes. El análisis de los SNP informativos mostró que entre los aislados españoles de M. bovis estudiados había un total de 1.345 SNP, de los cuales 773 eran singletones (presentes en un solo aislamiento). La diversidad genética entre los aislados era variable, con un número promedio de 62 SNP (rango = 49-88) desde que divergió de su antepasado común más reciente (MRCA). Los aislados se subdividieron en dos grupos distantes (A y B, Figura 2), con el más grande (A) subdividido en seis clades diferentes (A.1-A.3, A.4.1, A.4.2 y A.5) de acuerdo con BAPS y basado en una distancia máxima dentro de la cláusula de 57 SNP (Tabla complementaria S2; Figura 2; Figuras Todos los aislados del grupo A se caracterizaron por la presencia de dos SNP adicionales (ausentes en el grupo B) e incluían el 96 % (n = 95) del ganado y el 97,3 % (n = 36) de aislados de vida silvestre. El Grupo B incluía 5 aislados, uno de los cuales fue recuperado de un jabalí y el resto del ganado.
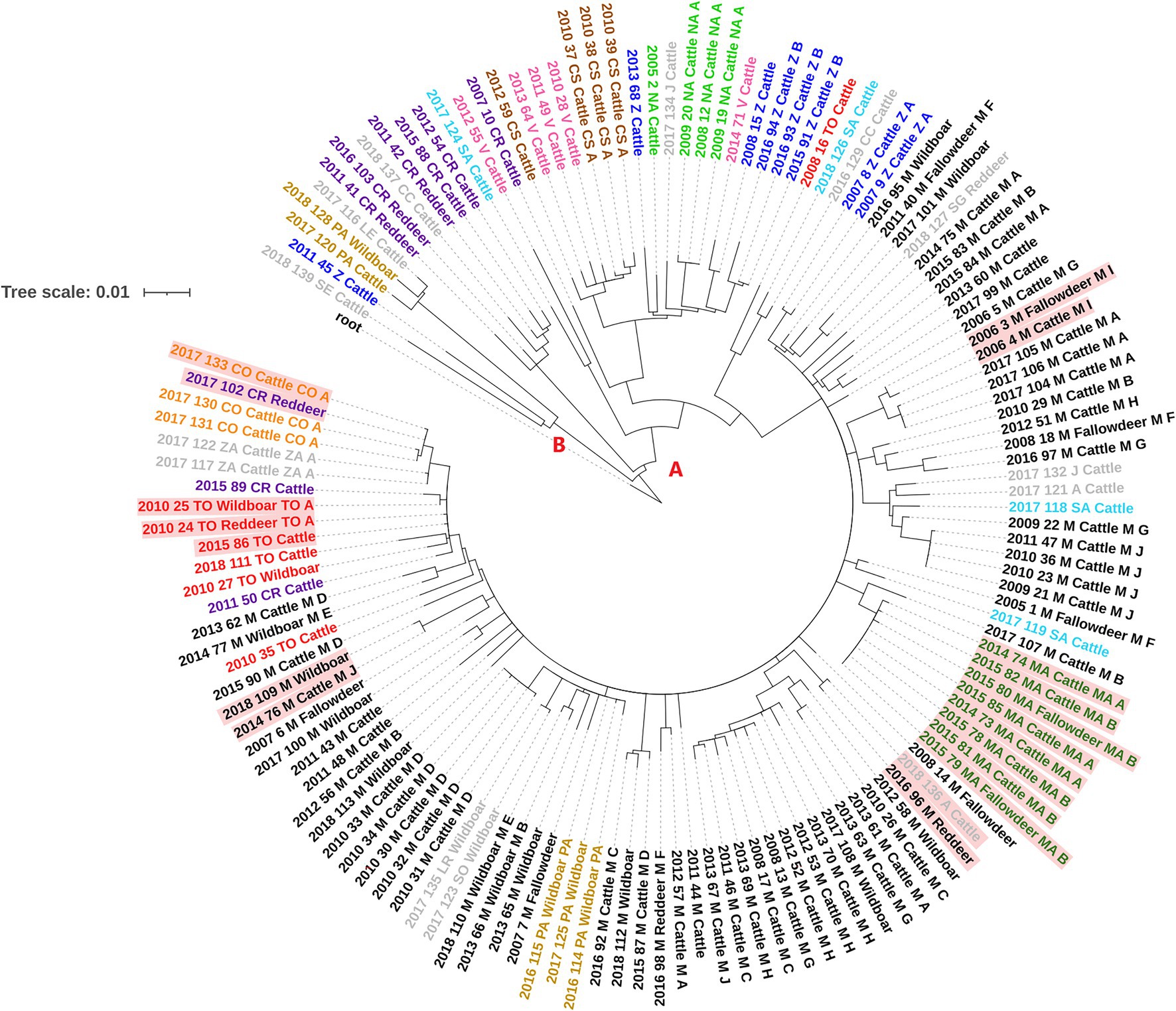 FIGURA 2. Árbol filogenético RAxML de secuencia de genoma completo construido utilizando un modelo GTR-CAT de 136 muestras españolas de M. bovis SB0339. Los dos grupos de aislados distantes se indican con las letras A y B. Las etiquetas de aislamiento son de color según la provincia de aislamiento, qué códigos se muestran en la Tabla 1. Los aislados con distancias genéticas de ≤3 SNP entre el ganado y la vida silvestre se colorean en un fondo rojo.
FIGURA 2. Árbol filogenético RAxML de secuencia de genoma completo construido utilizando un modelo GTR-CAT de 136 muestras españolas de M. bovis SB0339. Los dos grupos de aislados distantes se indican con las letras A y B. Las etiquetas de aislamiento son de color según la provincia de aislamiento, qué códigos se muestran en la Tabla 1. Los aislados con distancias genéticas de ≤3 SNP entre el ganado y la vida silvestre se colorean en un fondo rojo.
La distribución de los clados por provincia se muestra en la Figura Suplementaria S4. Clade A.2 representó el mayor clado/número de aislados (74,3 % del total) e incluyó todos los aislados de animales bovinos y de Madrid (n = 66) y Mallorca (n = 8), junto con 27 aislados de bovinos y vida silvestre de 12 provincias diferentes (Tabla complementaria S2; Figura complementaria S4). Los aislados de este clado se recuperaron durante todo el período de estudio (2005-2018) con un número medio de siete aislados por año (rango 1-21). Clade A.1 incluyó 3 aislados bovinos y 3 de vida silvestre recuperados entre 2011 y 2018 de provincias ubicadas en el centro y sur de España, mientras que los aislados de los clades A.3, A.4.1, A.4.2 y A.5 eran exclusivamente de ganado y se cultivaron en 2007, 2008, 2016 y 2018 (clade A.3, n = 4), Los aislados de los clados A.3, A.4.1 y A.4.2 se recuperaron de las provincias que cubrían la franja central de oeste a este de España y dos regiones del norte, mientras que los dos aislados en el clado A.5 se originaron de provincias muy lejanas (>600 km) (Figura complementaria S4). Finalmente, los aislados del grupo B se recuperaron en 2011 (n = 1) y 2017-2018 (n = 4).
La distancia genética entre el grupo (A frente a B) oscilaba entre 115 y 163 SNP, mientras que las distancias genéticas dentro del grupo eran mucho más bajas en el grupo A (mediana = 37 SNP, rango = 0-1229) que en el grupo B (mediana = 96 SNP, rango = 12-108, tabla complementaria S2). La distancia genética mediana dentro de las glomos en el grupo A fue de 30 (rango = 21-57; Tabla Suplementaria S2), mientras que la distancia genética mediana entre las glos fue de 85 (rango = 35-1414). Aunque los aislados del grupo B eran genéticamente distantes (distancia media 96 SNP), dos aislados de ganado y un jabalí estaban relativamente estrechamente relacionados (<16 SNP) y se recuperaron cerca en el tiempo (2017 y 2018).
Diversidad genética entre y dentro de las especificaciones
La diversidad genética entre aislados de diferentes huéspedes se evaluó utilizando 90 aislados (53 de ganado, incluido solo uno por rebaño, y todos los aislados de vida silvestre: 20 de jabalí, 9 de ciervos y 8 de ciervos rojos).
Se encontraron seis eventos de distancias genéticas cortas (≤3 SNP) entre aislados de diferentes especies huésped recuperadas cerca en el tiempo (dentro de 5 años, y dentro de <2 años en cinco de ellos) y espacio (dentro de la misma provincia o provincias vecinas). Específicamente, estos eventos incluyeron: un aislamiento de ganado y un ciervo rojo recuperados en Ávila y Madrid, respectivamente, un aislado de ganado de Córdoba y una muestra de ciervos rojos de Ciudad Real, un triplete formado por un ganado, un jabalí y un aislado de ciervo rojo M. bovis de Toledo, dos muestras de M. bovis aisladas de ganado y ciervos recuperados recuperado en Mallorca (Figura 2).
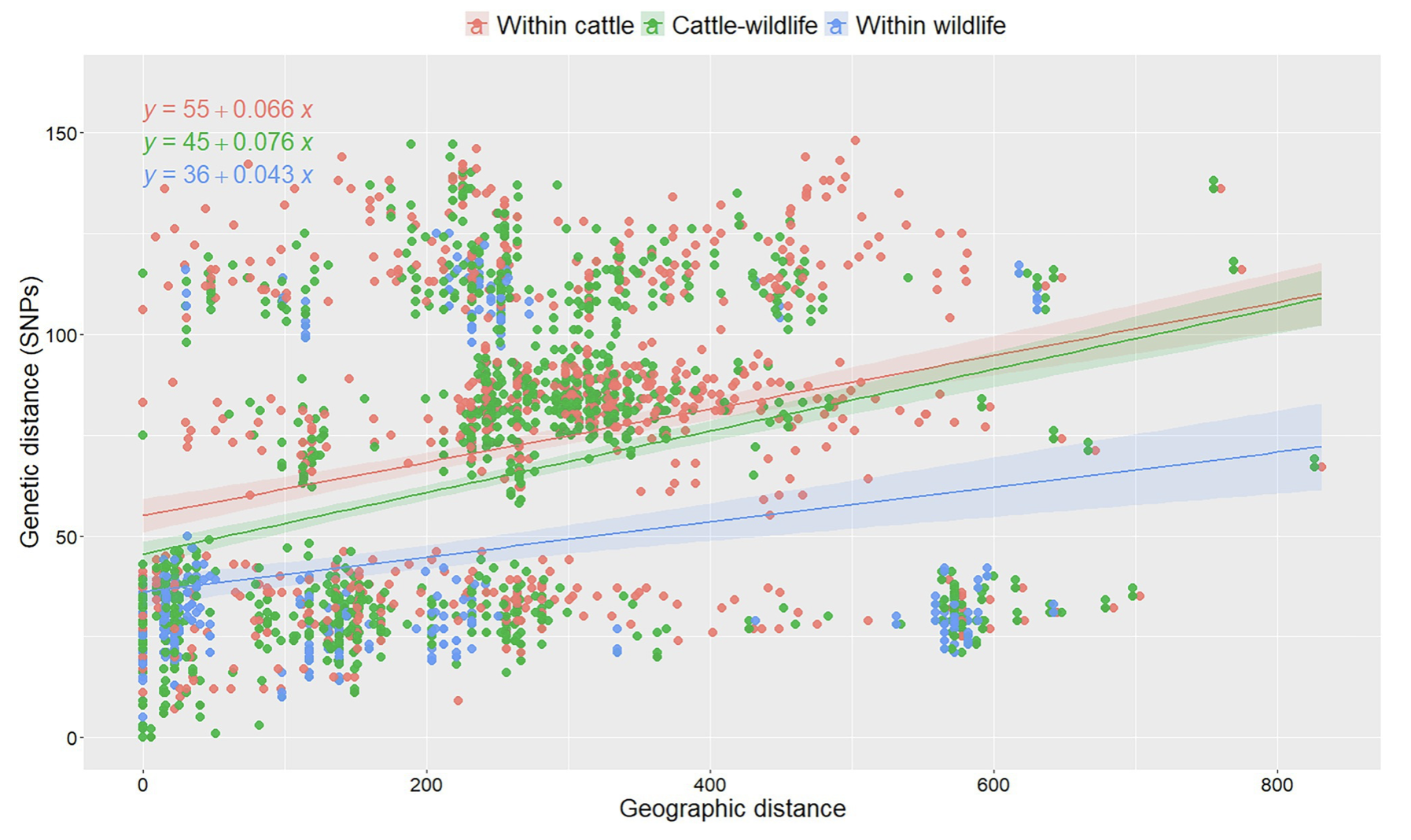
neral, la mediana de la divergencia genética dentro de los aislamientos de ganado en España (mediana = 80 SNP, IQR = 36–109), fue significativamente (p < 0,001, Kruskal-Wallis) más alta que entre el ganado y la vida silvestre (mediana = 66 SNP, IQR = 29– 93), y aislamientos dentro de la vida silvestre (mediana = 29 SNP, IQR = 22–40). No se observaron cambios cuando se volvió a evaluar la variabilidad dentro de la especie del ganado utilizando 10 selecciones aleatorias de aislamientos de los rebaños con múltiples aislamientos disponibles (cambio porcentual máximo 0,01%). Las distancias genéticas se correlacionaron de baja a moderada con las distancias geográficas (rho = 0.42, p < 0.001). Cuando se analizó la asociación entre las distancias genética y geográfica teniendo en cuenta el huésped del que se originaron los aislamientos utilizando un modelo lineal, tanto el huésped como la interacción entre el huésped y la distancia geográfica fueron significativos (Tabla complementaria S3): la distancia genética aumentó con el aumento de las distancias geográficas entre los ubicación de donde se originaron los dos aislados, pero este efecto fue menos pronunciado cuando se consideraron dos aislados de ganado en comparación con dos aislados de vida silvestre (y similar cuando se consideraron parejas recuperadas tanto de ganado como de vida silvestre; Figura 3). Se esperaba un aumento de 5 SNP en la diversidad genética dentro de los aislamientos de la vida silvestre por cada aumento de 100 km entre el origen de cada par de aislamientos, mientras que este número aumentó a 7 cuando ambos aislamientos se recuperaron del ganado (Tabla complementaria S3; Figura 3). Los coeficientes y las pendientes de los 10 modelos construidos con base en las diferentes selecciones aleatorias de aislamientos de ganado de rebaños infectados crónicamente fueron similares (<10% de cambio), lo que sugiere que la selección aleatoria de aislamientos en rebaños muestreados intensivamente no tuvo un impacto sustancial en la diversidad genética observada. (Figura 3; Figura complementaria S5; Tabla complementaria S3).
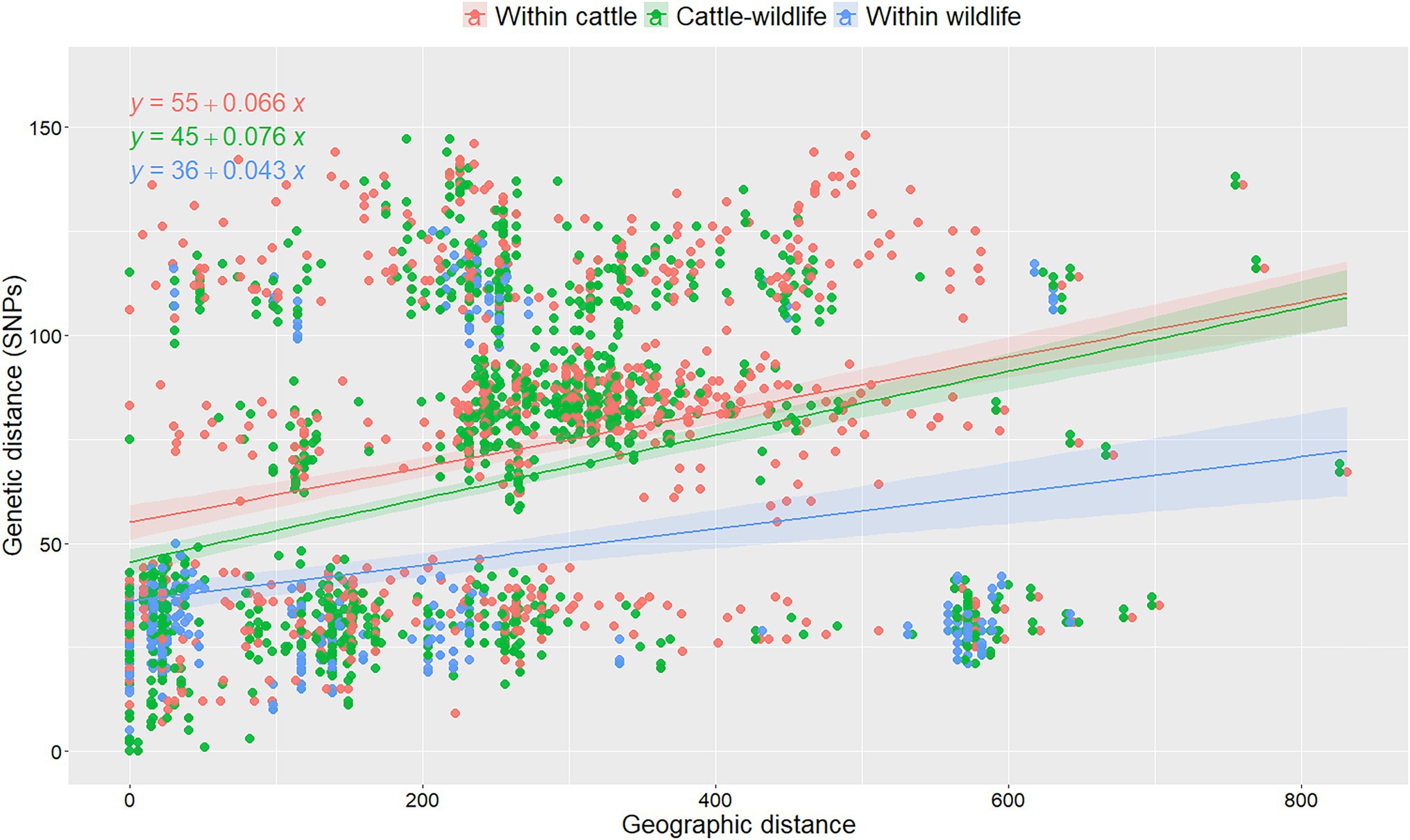 FIGURA 3. Análisis de las distancias genéticas por pares entre aislados recuperados dentro y entre especies en función de sus distancias geográficas de aislamiento en una de las 10 réplicas basadas en la selección aleatoria de un aislado por rehado infectado crónicamente. Se ajustaron líneas de regresión lineal para denotar la relación entre las distancias genéticas y geográficas por combinación de especies animales y se representaron fórmulas para cada categoría. Los colores denotan diferentes combinaciones de pares de aislados recuperados del ganado (rojo), vida silvestre (verde) y recogidos del ganado y la vida silvestre (azul). Las ecuaciones matemáticas se representaron para cada combinación de especies animales.
FIGURA 3. Análisis de las distancias genéticas por pares entre aislados recuperados dentro y entre especies en función de sus distancias geográficas de aislamiento en una de las 10 réplicas basadas en la selección aleatoria de un aislado por rehado infectado crónicamente. Se ajustaron líneas de regresión lineal para denotar la relación entre las distancias genéticas y geográficas por combinación de especies animales y se representaron fórmulas para cada categoría. Los colores denotan diferentes combinaciones de pares de aislados recuperados del ganado (rojo), vida silvestre (verde) y recogidos del ganado y la vida silvestre (azul). Las ecuaciones matemáticas se representaron para cada combinación de especies animales.
Por el contrario, cuando se realizó el mismo análisis en el subconjunto de aislados recuperados en Madrid, no se identificaron diferencias significativas (p > 0,05, prueba de Kruskal-Wallis) en las distancias encontradas entre los aislados de la misma especie (rango medio dentro del ganado = 30-32 SNP; mediana dentro de la vida silvestre = 30 SNP) o diferentes El modelo final incluyó solo la distancia geográfica entre los aislados como covariable, ya que no se encontró ningún efecto significativo de la especie animal. Se observó un aumento de 13 SNP por 100 km de distancia entre pares de aislados basado en los resultados de los 10 modelos de regresión lineal (Tabla complementaria S4). Los coeficientes de los modelos de regresión fueron más susceptibles a la variación basada en los aislados seleccionados al azar por rehada (hasta un cambio del 20 %, Tabla Suplementaria S4).
Diversidad genética dentro y entre el rebaño
Entre las 99 secuencias de ganado, 61 se originaron en los mismos 15 rebaños y fueron seleccionados para evaluar la diversidad dentro de los rebaños (Tabla 2). El número medio de aislados procedentes de las 15 rehadas fue de 3 (IQR = 3-5) y se recuperaron en una mediana de 2 años diferentes (IQR = 1-3). Diez de estas manadas se consideraron infectadas crónicamente (aislados recuperados en >1 año), produciendo 48 aislados de ganado (número medio de aislados por rebaño infectado crónicamente = 5, rango = 3-8) recuperados a lo largo de una mediana de 4 años diferentes (IQR = 2-5). Los 5 rehadas restantes (n = 5) no infectados crónicamente representaron 2-3 muestras recuperadas en un solo año (Tabla 2).
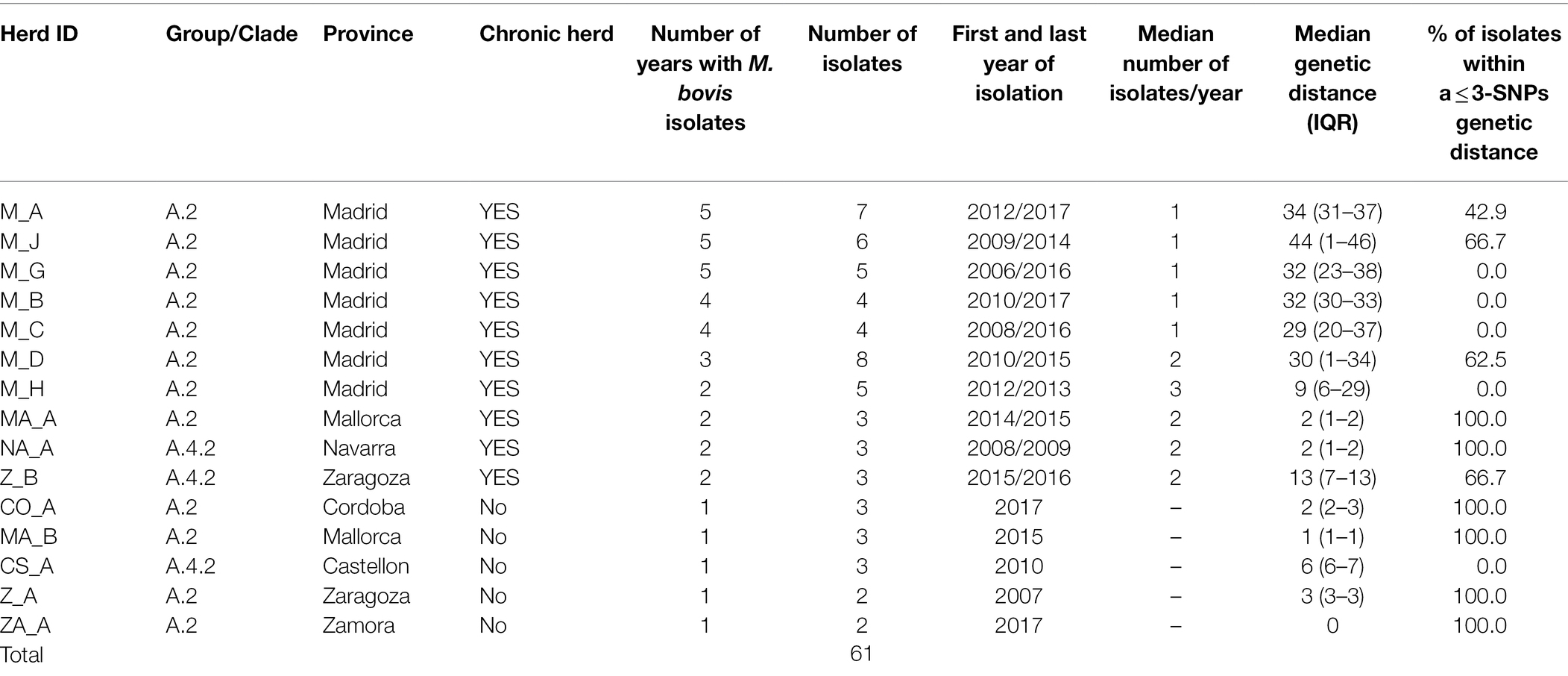 TABLA 2. Identificación del rebaño, provincia de origen, número de aislados, años entre el primer y el último aislamiento, número medio de aislados recuperados por año y % de aislados dentro de una distancia genética de ≤ 3-SNPs en rebaños incluidos en el análisis de la diversidad genética dentro del rebaño de Mycobacterium bovis.
TABLA 2. Identificación del rebaño, provincia de origen, número de aislados, años entre el primer y el último aislamiento, número medio de aislados recuperados por año y % de aislados dentro de una distancia genética de ≤ 3-SNPs en rebaños incluidos en el análisis de la diversidad genética dentro del rebaño de Mycobacterium bovis.
Los aislados de los análisis genéticos dentro del rebaño se incluyeron en tres clados del grupo A (A.2, A.4.1 y A.4.2). Las distancias genéticas medianas dentro del rebaño entre los aislados de cada uno de los 15 rebaños oscilaron entre 0 y 44 SNP (mediana = 9). Se observó una mayor diversidad genética en aislados recuperados de rehadas ubicadas en Madrid (a saber, M_A-M_C, M_G y M_H), aunque los aislados de rehadas individuales tendían a agruparse (Figura suplementaria S6). Los aislados recuperados en el retría CS_A, recuperados dentro del mismo año, estaban en un rango de 5-7 SNP separados. La distancia genética entre los aislados recuperados de los 10 rebaños infectados crónicamente fue de 2-4 SNP (mediana = 30), mientras que la diversidad genética fue menor en los aislados de los 5 rebaños restantes que no estaban infectados crónicamente con bTB (mediana de la distancia genética dentro del rebaño = 2 SNP, rango = 0-6).
En cinco de los 10 rehados infectados crónicamente, al menos el 60 % de los aislados recuperados en un rango de 2 a 5 años diferentes estaban a una distancia de 3 SNP entre sí (Tabla 2). De hecho, hubo una alta correlación positiva entre las distancias genéticas medias y el número de años diferentes a partir de los cuales se originaron los aislados en un reh (rho = 0,88, p < 0,001). Seis (M_A, M_C, M_D, M_H y M_J) de los siete rehados infectados crónicamente ubicados ubicados en Madrid incluyeron aislados similares (es decir, ≤3 SNP), casi similares (es decir, 4 SNP para M_C y M_H) y diferentes (es decir, >20Sin embargo, dos de estas manadas (M_D y M_H) produjeron aislados separados por distancias genéticas que oscilaron entre 29 y 37 SNP que se recuperaron en el mismo mes (Figura 4). Este patrón de distribución bimodal de las distancias genéticas por pares dentro del rebaño no fue evidente en el rebaño crónico restante de Madrid y en los rebaños crónicos ubicados en otras regiones (a saber, M_B, MA_A, NA_A y Z_B, Figura 4). Además, se identificó un evento de posible persistencia o reintroducción de cepas similares en el rehado M_G, como lo demuestran las cepas estrechamente relacionadas (6 SNP) en la manada encontrada con 5 años de diferencia (Figura 4).
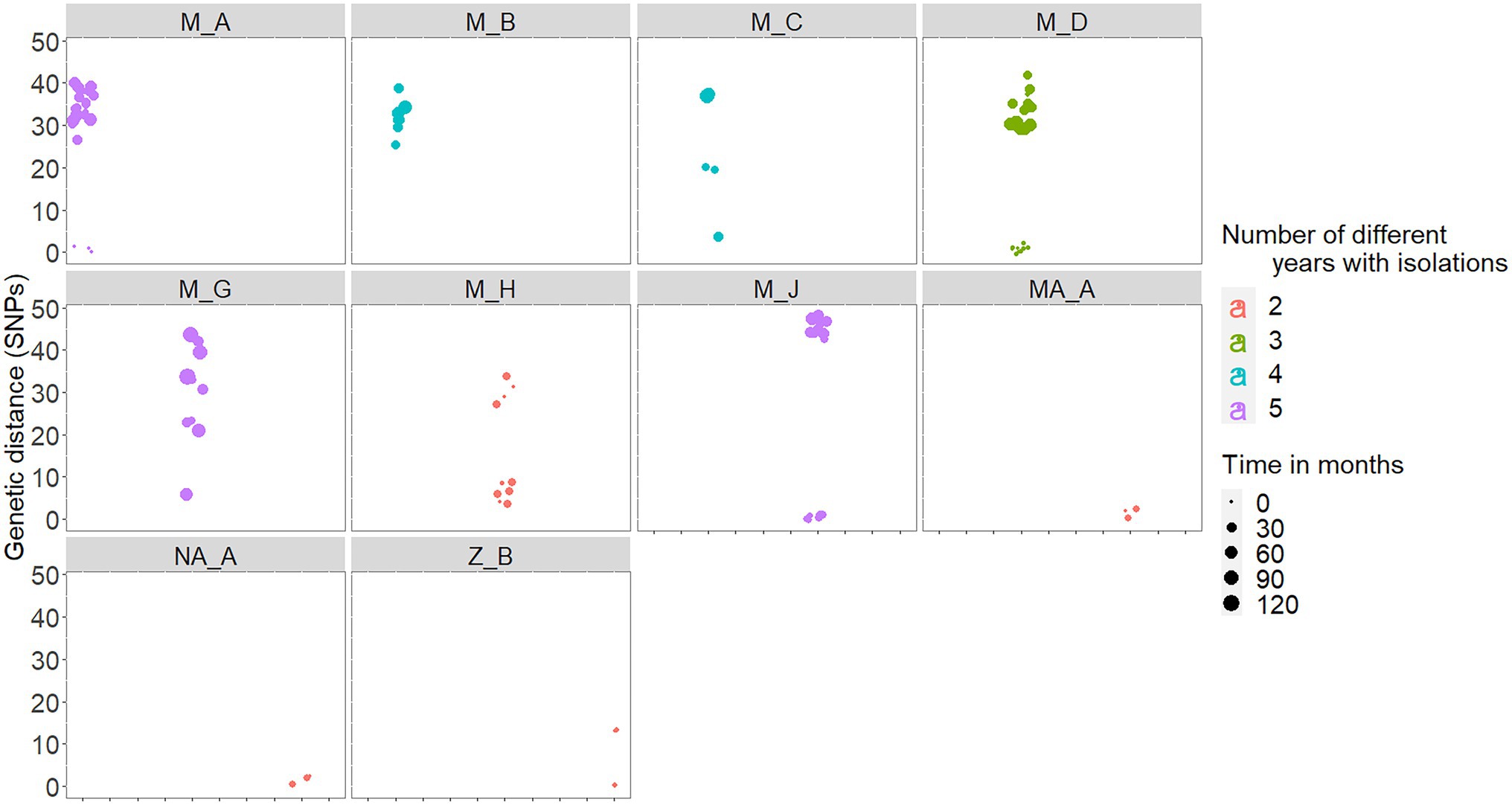 FIGURA 4. Distancias genéticas dentro del rebaño entre los aislados recuperados en los 10 rebaños infectados crónicamente. Las distancias genéticas se colorean en función del número de años diferentes con aislados de M. bovis (entre 2 y 5 años diferentes), y el tamaño representa el número de meses transcurridos entre cada par de aislados.
FIGURA 4. Distancias genéticas dentro del rebaño entre los aislados recuperados en los 10 rebaños infectados crónicamente. Las distancias genéticas se colorean en función del número de años diferentes con aislados de M. bovis (entre 2 y 5 años diferentes), y el tamaño representa el número de meses transcurridos entre cada par de aislados.
Discusión
En este estudio, la información del WGS de un gran panel de aislados de M. bovis recuperados de varias especies y regiones animales de España se utilizó para comprender el potencial de transmisión entre especies y dentro de las especies y para dilucidar el papel de la vida silvestre en la transmisión de bTB. Este es el primer estudio geómico a gran escala que describe la diversidad de M. bovis en la interfaz ganado-salvaje de múltiples provincias/regiones de España y destaca el papel relevante que los enfoques genómicas y filogenéticos pueden tener para adquirir conocimientos sobre la epidemiología de la bTB.
Seleccionamos los aislados de SB0339, ya que este fue el tercer tipo de spoligo más frecuente en España, y el más frecuentemente recuperado tanto en las poblaciones de ganado como en las de vida silvestre. Se ha aislado de forma recurrente, principalmente de las regiones centro-sur y norte del país, con evidencia de transmisión entre especies (Garcia-Jimenez et al., 2013). Aunque otros espoligotipos también se han encontrado repetidamente en la interfaz ganado-salvaje (por ejemplo, SB0121 y SB0134), más de un tercio de todos los aislados de SB0339 provienen de jabalí, ciervos rojos y ciervos (la mayor proporción de aislados de vida silvestre para un tipo de spoligo dado en España; mycoDB Además, SB0339 representa el patrón molecular aislado con más frecuencia en Madrid, incluyendo >50% de todas las muestras de M. bovis recuperadas en la región (mycoDB.es).
La proporción más alta (72,8 %) de aislados incluidos en este estudio se recuperaron del ganado, mientras que 37 aislados SB0339 se recuperaron de diferentes especies de vida silvestre. Más de la mitad de ellos (n = 20) fueron recuperados del jabalí, que se considera el principal anfitrión de mantenimiento en la Iberia mediterránea (Munoz-Mendoza et al., 2013; Gortazar et al., 2015). Los aislados incluidos en el estudio se tomaron muestras en diferentes momentos (datos heterocrónicas) entre 2005 y 2018. Este amplio intervalo de tiempo se seleccionó dada la pequeña tasa de mutación de M. bovis [estimaciones medias entre 0,15 y 0,5 eventos por genoma por año (Biek et al., 2012; Trewby et al., 2016; Crispell et al., 2017)], lo que hace necesario comparar los aislados recuperados a través de largos períodos al intentar reconstruir Del total de muestras secuenciadas, el 84,6 % (n = 115) se recuperaron después de 2009, ya que el ADN viable y de alta calidad era difícil de recuperar de las muestras (especialmente de la vida silvestre) recuperadas durante el período 2005-2009. No había aislamientos disponibles antes de 2005 en el Centro VISAVET.
Los datos de todo el genoma de los 136 aislados bTB revelaron una gran cantidad de SNP adquiridos dentro de los grupos A y B, con aislados de 47-72 y 38-61 SNP aparte de su MRCA, respectivamente (datos no mostrados). Además, se podían distinguir siete clados distintos entre la colección secuenciada, de los cuales tres contenían secuencias de vida silvestre y de M. bovis derivadas del ganado. Estos resultados pueden no solo ser sugestivos de una larga historia de endémica, sino también que la infección por M. bovis se ha transmitido entre las poblaciones de ganado y vida silvestre en España durante mucho tiempo. De hecho, se esperaba un alto grado de diversidad genética en los aislados SB0339, dado que representa el 8% de todos los aislados de M. bovis tipados en España, y ha estado circulando en varias áreas de España durante largos períodos de tiempo (Rodriguez-Campos et al., 2010; Gortazar y Boadella, 2014).
Las distancias genéticas entre pares entre los aislados en el clado principal A.2 fueron las más bajas de todos los clados, a pesar de que incluían el mayor número de secuencias (74,3% de toda la colección) e incluían aislados tanto del ganado como de la vida silvestre recuperada durante un largo período de tiempo. La similitud entre las cepas de este clado recuperadas de diferentes especies huésped es sugerente de eventos de transmisión recientes y/o transmisión desde una fuente común (no muestreada). Se ha informado evidencia de la transmisión de M. bovis entre el ganado y la vida silvestre en numerosos estudios en todo el mundo, especialmente en áreas donde coexisten varias especies susceptibles. Un estudio que incluyó un total de 27 spoligotipos diferentes realizados en Cataluña, una región ubicada en el noreste de España, reveló la transmisión entre diferentes huéspedes (Perea et al., 2021). La investigación realizada en el Reino Unido mostró que los aislados de tejón estaban en un rango de 0 a 4 SNPs aparte del aislado de ganado más cercano, y que la transmisión se produjo con más frecuencia de tejón a ganado que viceversa (Biek et al., 2012; Crispell et al., 2019). Utilizando un enfoque bayesiano, un estudio reciente realizado en Francia reveló una alta tasa de transmisión entre especies entre poblaciones de ganado y tejón (Duault et al., 2022). Además, también se sugirió una alta tasa de intercambio genético entre el ganado muestreado y las poblaciones de vida silvestre en Nueva Zelanda (Crispell et al., 2017).
En general, el tiempo y el origen geográfico de los aislados fueron buenos predictores de distancias genéticas, independientemente de la especie huésped de origen del aislado. La heterogeneidad genética era geográfica en lugar de específica de la especie huésped, ya que los aislados recuperados de diferentes especies animales dentro de las mismas provincias tendían a estar más estrechamente relacionados que los que se originaron en las mismas especies y diferentes provincias. Sin embargo, a pesar del potencial de transmisión entre especies sugerido por los hallazgos de nuestro estudio, la agrupación general de aislados por especies huésped señaló que las cepas de M. bovis recuperadas de los huéspedes salvajes eran menos diversas que las procedentes del ganado, y que las distancias genéticas estaban asociadas con la provincia de origen (Figura 2; Tabla Suplementaria S2). Esto fue particularmente evidente en el caso de las muestras de jabalí, con aislados recuperados de las mismas provincias o vecinas a pocos SNP de distancia entre sí. De hecho, un estudio anterior realizado en Cataluña sugirió que la proximidad y el vecindario eran los dos factores de riesgo más importantes asociados con la heterogeneidad genética observada (Perea et al., 2021). En nuestro estudio, se observó una heterogeneidad sustancialmente mayor en las distancias genéticas por pares entre los aislados de ganado en España en comparación con los aislados de vida silvestre y los pares de ganado y vida silvestre, lo que podría deberse a la transmisión de un grupo más grande de cepas SB0339 en el reservorio de ganado que no llegan a la vida silvestre (Figura Por el contrario, en el análisis de las cepas de Madrid, la diversidad genética de los aislados recuperados del ganado y de la vida silvestre fue más similar. Aunque se identificó una homogeneidad genética sustancial a nivel spoligo-VNTR entre los aislados de ganado en Madrid en un estudio anterior (de la Cruz et al., 2014), los análisis de todo el genoma realizados aquí revelaron un alto grado general de heterogeneidad genética independientemente de la especie de origen del huésped, lo que sugiere una larga historia de circulación endémica SB Sin embargo, no había información disponible sobre los perfiles de VNTR para este estudio, ya que la mecanografía de VNTR ya no se realiza de forma rutinaria en nuestro laboratorio. A pesar de que se identificó una relación significativa entre la distancia genética y geográfica de M. bovis en la región, la falta de asociación de la especie huésped con la distancia genética que separa dos aislados recuperados de Madrid también puede ser sugerente de la evolución de cepas en huéspedes no muestreados no considerados aquí (que podrían incluir poblaciones de ganado sin enlace de transmisión con la vida silvestre, Por el contrario, si solo se consideraron en el análisis provincias con una gran proporción de aislados de ganado y vida silvestre (es decir, Madrid, Ciudad Real y Toledo), los resultados obtenidos fueron los mismos que los del análisis general de España (datos no mostrados). Este análisis confirmó que, incluso después de realizar una comparación más similar de la diversidad genética en regiones con un mayor número de aislamientos de vida silvestre, Madrid es representativo de un escenario de transmisión SB0339 complicado e interespecífico. Además, la diversidad genética general de los aislados incluidos en el clado A.2 (Figuras Suplementarias S2, S4) reveló resultados similares a los de Madrid, ya que no se identificaron diferencias significativas en las distancias encontradas entre los aislados de la misma especie (rango medio dentro del ganado = 31-32 SNP; mediana dentro de la vida silvestre = 27 SNP La gran heterogeneidad (rango 55-163 SNP) identificada cuando se comparó el clado A.2 con los clados restantes identificados en regiones distintas de Madrid (Tabla Suplementaria S2) está de acuerdo con la hipótesis de una larga historia de endémica de este tipo de spoligo establecida en este estudio. Sin embargo, las relaciones filogenéticas descritas en este estudio pueden ser particularmente sensibles al sesgo de selección en la población muestreada de Madrid frente a los aislados secuenciados generales recuperados en múltiples provincias de España, y las diferencias en el efecto observado explicado aquí deben interpretarse con precaución. A pesar de la asociación observada entre distancias genéticas y geográficas, en varios casos se recuperaron aislados genéticamente no relacionados (es decir, muy distintos) de lugares cercanos (y en algunos casos incluso de la misma manada), lo que sugiere que la infección podría haber ocurrido en un lugar diferente, particularmente en el ganado, ya que la mayoría (>81%) de los aislados de Desafortunadamente, los movimientos individuales de todo el ganado muestreado no estaban disponibles para este estudio y, por lo tanto, no se pudo confirmar la hipótesis de nuevas introducciones debido al movimiento de animales infectados.
Además, tres de los seis grupos de cepas muy similares (≤3 SNP) recuperadas del ganado y la vida silvestre se encontraron en Madrid (Figura 2; Figura complementaria S2). De hecho, el patrón SB0339 se ha encontrado de forma recurrente en determinadas zonas, especialmente en el Parque Natural del Monte de El Pardo ubicado en el municipio de Madrid y en Colmenar Viejo, donde factores de riesgo como la gestión extensiva de los rebaños y la abundante presencia de reservorios de fauna pueden explicar las interespecies observadas. similitud genética y que la prevalencia de la TBb se ha mantenido históricamente alta tanto en el ganado como en la vida silvestre (Aranaz et al., 2004; Rodriguez-Campos et al., 2010).
Los análisis de la diversidad genética dentro del rebaño en aislados de rebaños infectados crónicamente tuvieron como objetivo proporcionar cierta información sobre el grado de diferencia esperada a lo largo del tiempo en un rebaño dado, lo que podría ayudar a diferenciar la fuente de un brote (es decir, recaída frente a la reinfección; Hatherell et al., 2016). Aquí se utilizó un umbral de ≤ 3 SNP para definir grupos de transmisión en los que estamos seguros de que cepas similares están circulando en diferentes huéspedes. Este valor fue inferior al seleccionado en el estudio realizado en Cataluña, donde se consideró una distancia por pares de ≤12 SNPs para definir grupos de transmisión putativos basados en los patrones de agrupación observados (Perea et al., 2021). En nuestro estudio, los aislados con ≤3 SNP se consideraron estrechamente relacionados porque la mayoría de los aislados epidemiológicamente vinculados recuperados de la misma manada y muestreados dentro del mismo año no superaron este nivel de divergencia. Cuando se aplicó un umbral de ≤ 10 SNPs en nuestros análisis, 2 rehadas adicionales (a saber, CS_A y M_H) de las 15 rehadas analizadas tenían >50% de las cepas muestreadas dentro de este corte circulando durante el período muestreado (Figura 4). El uso de diferentes umbrales puede afectar a las inferencias sugeridas aquí (es decir, recaída frente a nuevas presentaciones). Sin embargo, nuestro objetivo aquí era identificar grupos de patrones moleculares que probablemente hayan surgido de los vínculos epidemiológicos en lugar de la adquisición de SNP derivados del proceso de evolución. Además, seleccionamos este duro límite, ya que coincidió con el derivado de la revisión ampliada de la literatura del MTBC para identificar con precisión los eventos de transmisión, recaída y reinfecciones recientes (Kato-Maeda et al., 2013; Roetzer et al., 2013; Lee et al., 2015). En general, identificamos una fuerte relación de tiempo y origen espacial y variabilidad genética, de modo que esos factores fueron muy informativos para caracterizar la circulación, ya que los aislados tendían a agruparse en un patrón dentro del rebaño. Las distancias genéticas observadas tanto entre los rebaños como dentro de los rebaños fueron muy heterogéneas dependiendo de la historia epidemiológica (número de años con aislamientos de bTB en la manada), ya que las distancias genéticas medianas por pares observadas en los 10 rebaños muestreados crónicamente variaron en gran medida (rango 2-4 SNP) en comparación Esto se esperaba, ya que las manadas no infectadas crónicamente se tomaron muestras durante un período de 1 año frente a un número medio de 3,5 años diferentes con aislamientos en rejas infectadas crónicamente. Sin embargo, en la mitad de los rederos infectados crónicamente, la mayoría de los aislados recuperados durante varios años estaban separados por ≤ 3-SNP. La limitada diversidad genética dentro del rebaño que se encuentra de forma recurrente en varios rebaños muestreados, en muchas ocasiones a pesar del tiempo considerable transcurrido entre sus aislamientos, sugiere que los animales infectados crónicamente en el rebaño (que se habrían perdido en las pruebas de rebaño bTB) pueden haber contribuido a la persistencia de la enfermedad (Guta et al. Además, la agrupación genética de cepas procedentes de los mismos rehados (y mismas provincias) indicó que M. bovis circulaba continuamente en los rehados muestreados (sin la introducción de nuevas cepas). Alternativamente, la persistencia de cepas muy similares también puede sugerir reintroducciones de otras fuentes, como rehadas vecinas, persistencia ambiental o huéspedes alternativos, como se sugiere en otros lugares (Biek et al., 2012).
La diversidad genética observada en los aislados de M. bovis analizados por SB0339 no fue aparentemente causada por las recientes introducciones exógenas en España. En cambio, las distancias genéticas observadas sugirieron una infección endémica de automantenimiento dentro de diferentes especies animales en cada región, con ciertos eventos de transmisión entre especies, lo que fue particularmente evidente en el caso de Madrid. A pesar del pequeño tamaño de la muestra incluido en este estudio, este cuadro está de acuerdo con los hallazgos de estudios anteriores en España y en otros lugares sobre el efecto potencial de los factores de riesgo locales (es decir, proximidad espacial, gestión extensa en rejas de carne de vacuno, contacto con otras fuentes de infección) en áreas endémicas de bTB (Green et al., Además, nuestros resultados están de acuerdo con un estudio anterior realizado en Portugal, donde la diversidad genética observada apoyó la circulación natural de M. bovis durante mucho tiempo con múltiples interacciones de diferentes especies huésped (Reis et al., 2021). La interacción con los reservorios de vida silvestre se identificó como la segunda causa más importante de colapsos de la manada en España, después de la infección residual (Guta et al., 2014a). De hecho, los aislados de M. bovis recuperados de especies de vida silvestre en el Parque Nacional de Doñana eran de hecho más prevalentes en el ganado, contribuyendo así a la persistencia de bTB (Romero et al., 2008). El ganado y los tejones encontrados en el norte de España en la misma área geográfica y durante el mismo período compartieron espoligotipos similares, lo que sugiere una dinámica común de la infección (Balseiro et al., 2013). Teniendo todo en cuenta, la mayor diversidad de huéspedes identificada en la epidemiología de bTB en España conduce a cadenas de transmisión más crecientes y diversas (Barasona et al., 2019). También se identificaron vínculos epidemiológicos entre diferentes huéspedes que involucran a la misma especie analizada aquí (es decir, jabalí y ciervos rojos) en países vecinos como Portugal (Cunha et al., 2012; Reis et al., 2020) y Francia (Hauer et al., 2015; Michelet et al., 2019), aunque se identificaron diferentes patrones moleculares. Los resultados obtenidos aquí son importantes teniendo en cuenta el grado relativamente alto de persistencia que existe en varias regiones de España, como las partes central y suroeste del país.
El muestreo de diferentes especies huésped realizadas aquí coincidió en unidades temporales (años) y espaciales (provincia de origen) similares. Sin embargo, debido a la fuerte dependencia de la disponibilidad de aislamiento en la recolección de cepas VISAVET y al grado moderado de éxito (~60%) en la recultivo de los aislados, los sesgos incluidos en este estudio fueron inevitables.
El potencial de los análisis derivados de WGS puede verse comprometido cuando una población huésped mal muestreada contribuye a la transmisión, que en este caso podrían ser los reservorios de vida silvestre. Además, el muestreo de la población huésped a menudo se limita a las investigaciones de brotes en las que el muestreo se reduce a los rebaños afectados y la inclusión de otras fuentes, como granjas cercanas o reservorios de vida silvestre, enfrenta limitaciones financieras, éticas o logísticas. Más de la mitad de los aislamientos de fauna silvestre incluidos aquí (n = 20) se originaron a partir de muestras recolectadas de jabalíes, principalmente de Madrid, y la mayoría (n = 15/17) de los aislamientos de M. bovis de ciervo y gamo se muestrearon en las regiones centrales del país también. Por el contrario, otras provincias estaban claramente infrarrepresentadas en nuestra colección: el centro de España recibió un muestreo más intensivo en comparación con las regiones del sur, donde la tuberculosis bovina es muy frecuente tanto en el ganado como en la vida silvestre (Ministerio de Agricultura, Pesca, Alimentación y Medio Ambiente, 2020; Ministerio de Agricultura , Pesca y Alimentación, 2021). El esfuerzo de muestreo diferencial entre regiones puede haber afectado la solidez de nuestros hallazgos, particularmente con respecto a la vida silvestre, al considerar todo el país. Por lo tanto, nuestros hallazgos no deben extrapolarse a otras áreas a menos que estudios adicionales confirmen el grado de diversidad dentro y entre las especies que se encuentran aquí. No obstante, los aislados pertenecientes al espoligotipo SB0339 proceden mayoritariamente de Madrid, que fue la región más intensamente muestreada y en la que se consiguió un mejor equilibrio ganado-fauna (49% del total de aislados, relación ganado:fauna MADRID 2:1 Vs. ganado: vida silvestreOVERALL 3:1). Con respecto al ganado, se implementaron estrategias similares de prueba y sacrificio basadas en pruebas anuales utilizando la prueba única de tuberculina intradérmica en todas las provincias incluidas aquí durante el período de estudio y, por lo tanto, una representación razonable de ganado bTB positivo infectado con SB0339 circulando en España. estaba disponible. Aunque el control y la vigilancia de la TBb en la vida silvestre no están tan estandarizados como en el ganado bovino y había menos aislamientos de vida silvestre disponibles para el análisis, se recuperó una gran fracción de aislamientos de vida silvestre de Madrid (con una alta prevalencia de TBb y densidad de vida silvestre), lo que sugiere que los resultados observado aquí puede ser una buena aproximación del escenario real de SB0339 no solo en el ganado, sino también en la vida silvestre (Ministerio de Agricultura, Pesca, Alimentación y Medio Ambiente, 2020; Ministerio de Agricultura, Pesca y Alimentación, 2021).
Además, entre las 136 muestras, había 7 aislados con baja (<20x) profundidad de cobertura, con regiones de ADN de poca o nula cobertura que llevaron a la identificación de SNP poco fiables. Del mismo modo, el porcentaje del genoma de referencia cubierto por estas secuencias estaba por debajo del 99 %, lo que para los organismos clonales como M. bovis, implica la necesidad de una cantidad significativa de corrección. Aunque los resultados obtenidos en estas muestras de baja cobertura deben considerarse con precaución, no se esperaba ningún sesgo importante debido a las llamadas erróneas de SNP. Estas llamadas ambiguas ocurrieron en SNP no informativos (SNP compartidos por todos los aislados incluidos en el estudio que no eran relevantes para definir el patrón de agrupación presentado aquí). Las llamadas erróneas se verificaron meticulosamente con IGV, y los SNP confiables se corrigieron manualmente como sugiere la documentación del vSNP (Orloski et al., 2018) y se realizaron en varios estudios (Orloski et al., 2018; Salvador et al., 2019; Perea et al., 2021; Reis et al., 2021 En caso de que se presentaran llamadas erróneas en áreas con baja cobertura o problemas de mapeo, se filtraron manualmente.
Los métodos de genotipado de alto rendimiento, como el WGS, han creado oportunidades sin precedentes para estudiar la red de transmisión de microorganismos como M. bovis y permiten rastrear las fuentes de infección, lo que puede complementar otras medidas incluidas en el programa de erradicación de bTB en España. Dada la creciente rentabilidad de las técnicas de caracterización basadas en WGS, creemos que la tipificación basada en WGS eventualmente se convertirá en el estándar para los estudios epidemiológicos moleculares de bTB, al igual que también ha sido la tendencia con otros patógenos (por ejemplo, patógenos transmitidos por alimentos). Este estudio confirmó que M. bovis probablemente se mantiene en poblaciones multi en lugar de solo huésped en áreas de alta pero también baja prevalencia (por ejemplo, Mallorca), y que la contribución relativa de los reservorios de vida silvestre al mantenimiento de bTB en algunas regiones puede ser baja en comparación con el centro y suroeste de España. Si bien las técnicas de mecanografía tradicionales han demostrado que los patrones moleculares de M. bovis se mantienen dentro de cúmulos espaciales bien definidos, su poder para discriminar aún más las cepas dentro de los cúmulos, lo que podría ayudar a explicar la persistencia y la transmisión, es limitado (Trewby et al., 2016). En este sentido, el WGS es una herramienta valiosa para mejorar la comprensión de la epidemiología de bTB, incluso para patógenos que evolucionan lentamente y se conservan genéticamente como M. bovis (Garnier et al., 2003). Aquí, el WGS se utilizó para describir la heterogeneidad genética en un espoligotipo altamente predominante en un intento de evaluar el potencial de transmisión entre especies independientemente de la dirección, ya que los árboles filogenéticos presentados aquí no son equivalentes a los árboles de transmisión. La combinación de datos de la secuencia de M. bovis y modelos matemáticos que consideran la estructura temporal inherente a las muestras heterocrónicas seleccionadas puede aumentar el poder estadístico para inferir los procesos evolutivos de M. bovis, como se realizó en investigaciones anteriores (Glaser et al., 2016; Crispell et al., 2017, 2019; Salvador et al., 2019). La inclusión de un reloj molecular en los análisis realizados aquí y la adición de una selección equilibrada de muestras entre el ganado y la vida silvestre serán objeto de los siguientes análisis. En última instancia, la adición de la escala temporal en el análisis de las heterogeneidades genéticas entre los aislados puede ayudar a cuantificar el papel de los reservorios de vida silvestre y el ganado en la dinámica de la infección por M. bovis en España.
Declaración de disponibilidad de datos
Los conjuntos de datos generados y analizados para este estudio se pueden encontrar en el Archivo de Lectura de Secuencia de Información del Centro Nacional de Biotecnología (NCBI-SRA) y se puede acceder con el número de Bioproyecto PRJNA804719.
Declaración ética
No se requirió la revisión y aprobación ética para el estudio de los animales porque todas las muestras se recogieron como parte de la vigilancia regulatoria autorizada en el marco del Programa Nacional de Erradicación de la Tuberculosis Bovina de España. No se obtuvo el consentimiento informado por escrito para la participación de los propietarios porque no se requería la aprobación de los propietarios de los locales para este estudio.
Contribuciones del autor
PP y JA diseñaron el estudio, escribieron el manuscrito y recibieron comentarios sustanciales de todos los autores. PP y VL-L realizaron los cultivos bacteriológicos de las muestras y la extracción de ADN. PP analizó los datos con la ayuda de JA, VL-L, SR-A, JH, TS, BR, JB y LJ. JLS proporcionó asesoramiento sobre la interpretación de los datos. Todos los autores contribuyeron al artículo y aprobaron la versión enviada.
Colaboradores de la Red Española de Vigilancia y Monitoreo de la Tuberculosis Animal
Los miembros de la Red Española de Vigilancia y Monitoreo de la Tuberculosis Animal que participaron en este estudio incluyen el Ministerio Español de Agricultura, Pesca y Alimentación junto con los Laboratorios Centrales y Regionales; Laboratorio Central de Sanidad Animal de Santa Fe, Granada; Laboratorio Regional de Sanidad Animal, Junta de Castilla y León; Instituto de Biología Animal Balears; Instituto de Investigaciones en Recursos Cinegéticos (IRE Ganadería y Protección Animal, Subdirección General de Higiene y Seguridad Alimentaria, Comunidad de Madrid.
Financiación
Esta investigación fue una contribución al proyecto Estrategias Integradas para el Control y la Erradicación de la Tuberculosis en España (ERATUB; RTI2018-096010-B-C22, Ministerio de Ciencia, Innovación y Universidades). El Ministerio de Agricultura, Pesca y Alimentación y el Área de Ganadería de la Comunidad de Madrid apoyaron esta publicación. JA fue el destinatario de un contrato de Ramón y Cajal de MINECO (RYC-2016-20422).
Conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un posible conflicto de intereses.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o reclamación que pueda ser hecha por su fabricante, no está garantizado ni respaldado por el editor.
Agradecimientos
Queremos dar las gracias a Francisco Lozano y Alexandra Gutiérrez por su apoyo durante los análisis de laboratorio; al Ministerio de Agricultura, Pesca y Alimentación de España, a la Comunidad Autónoma de Madrid, al Laboratorio Central de Sanidad Animal (LCSA, Santa Fe-Granada); y a los Gobiernos Regionales de Castilla y León por proporcionar muestras incluidas en el estudio.
Material complementario
El material complementario de este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fmicb.2022.915843/full#supplementary-material
Notas al pie de página
Referencias
Allen, A. R., Skuce, R. A., y Byrne, A. W. (2018). Tuberculosis bovina en Gran Bretaña e Irlanda – ¿Una tormenta perfecta? La confluencia de posibles impedimentos ecológicos y epidemiológicos para controlar una enfermedad infecciosa crónica. Delante. Veterinario. Ciencia. 5:109. doi: 10.3389/fvets.2018.00109
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Aranaz, A., De Juan, L., Montero, N., Sanchez, C., Galka, M., Delso, C., et al. (2004). Tuberculosis bovina (Mycobacterium bovis) en la vida silvestre en España. J. Clin. Microbiol. 42, 2602–2608. doi: 10.1128/JCM.42.6.2602-2608.2004
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Balseiro, A., Gonzalez-Quiros, P., Rodriguez, O., Francisca Copano, M., Merediz, I., de Juan, L., et al. (2013). Relaciones espaciales entre los tejones euroasiáticos (Meles meles) y el ganado infectado con Mycobacterium bovis en el norte de España. Veterinario. J. 197, 739-745. doi: 10.1016/j.tvjl.2013.03.017
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Barasona, J. A., Gortazar, C., de la Fuente, J., y Vicente, J. (2019). La riqueza de los anfitriones aumenta el riesgo de enfermedad de tuberculosis en las áreas gestionadas por el juego. Microorganismos 7:60182. doi: 10.3390/microorganismos7060182
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Biek, R., O’Hare, A., Wright, D., Mallon, T., McCormick, C., Orton, R. J., et al. (2012). La secuenciación del genoma completo revela patrones de transmisión local de Mycobacterium bovis en poblaciones de ganado simpátricos y tejones. PLoS Pathog. 8:e1003008. doi: 10.1371/journal.ppat.1003008
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Bivand, R., Keitt, T., y Rowlingson, B.. (2013). rgdal: Enlaces para la Biblioteca de Abstracción de Datos «Geoespaciales». Paquete R versión 1.4-8.
Cheng, L., Connor, T. R., Sirén, J., Aanensen, D. M., y Corander, J. (2013). Agrupación jerárquica y espacialmente explícita de secuencias de ADN con software BAPS. Mol. Biol. Evol. 30, 1224–1228. doi: 10.1093/molbev/mst028
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Descifrando la biología de Mycobacterium tuberculosis a partir de la secuencia completa del genoma. Naturaleza 393, 537-544. doi: 10.1038/31159
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Comas, I., Homolka, S., Niemann, S., y Gagneux, S. (2009). Genotipado de bacterias genéticamente monomórficas: la secuenciación de ADN en Mycobacterium tuberculosis destaca las limitaciones de las metodologías actuales. PLoS One 4:e7815. doi: 10.1371/journal.pone.0007815
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Primos, D. V. (2001). Infección y control de Mycobacterium bovis en el ganado doméstico. Rev. Ciencia. Tecnología. 20, 71-85. doi: 10.20506/rst.20.1.1263
Primos, D. V., Wilton, S. D., y Francis, B. R. (1991). Uso de la amplificación de ADN para la rápida identificación de Mycobacterium bovis. Veterinario. Microbiol. 27, 187–195. doi: 10.1016/0378-1135(91)90010-D
Crispell, J., Benton, C. H., Balaz, D., De Maio, N., Ahkmetova, A., Allen, A., et al. (2019). Combinando genómica y epidemiología para analizar la transmisión bidireccional de Mycobacterium bovis en un sistema multihost. elife 8:45833. doi: 10.7554/eLife.45833
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Crispell, J., Zadoks, R. N., Harris, S. R., Paterson, B., Collins, D. M., de-Lisle, G. W., et al. (2017). Uso de la secuenciación del genoma completo para investigar la transmisión en un sistema multihost: tuberculosis bovina en Nueva Zelanda. BMC Genomics 18:180. doi: 10.1186/s12864-017-3569-x
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Cunha, M. V., Matos, F., Canto, A., Albuquerque, T., Alberto, J. R., Aranha, J. M., et al. (2012). Implicaciones y desafíos de la tuberculosis en los ungulados de vida silvestre en Portugal: una perspectiva de epidemiología molecular.Res. Veterinario. Ciencia. 92, 225–235. doi: 10.1016/j.rvsc.2011.03.009
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
de la Cruz, M. L., Pérez, A., Bezos, J., Pages, E., Casal, C., Carpintero, J., et al. (2014). Dinámica espacial de la tuberculosis bovina en la Comunidad Autónoma de Madrid, España (2010-2012). PLoS One 9:e115632. doi: 10.1371/journal.pone.0115632
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
de la Rua-Domenech, R., Goodchild, A. T., Vordermeier, H. M., Hewinson, R. G., Christiansen, K. H., y Clifton-Hadley, R. S. (2006). Diagnóstico ante mortem de tuberculosis en ganado: una revisión de las pruebas de tuberculina, ensayo de gamma-interferón y otras técnicas de diagnóstico auxiliar. Res. Veterinario. Ciencia. 81, 190–210. doi: 10.1016/j.rvsc.2005.11.005
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Duault, H., Michelet, L., Boschiroli, M.-L., Durand, B., y Canini, L. (2022). Un modelo evolutivo bayesiano para comprender la contribución de la vida silvestre a la transmisión de la familia F4 Mycobacterium bovis en el suroeste de Francia. Veterinario. Res. 53:28. doi: 10.1186/s13567-022-01044-x
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Panel de la EFSA sobre Salud y Bienestar Animal (AHAW) (2014). Declaración sobre un marco conceptual para la tuberculosis bovina. EFSA J. 12:59. doi: 10.2903/j.efsa.2014.3711
García-Jimenez, W. L., Fernández-Llario, P., Benitez-Medina, J. M., Cerrato, R., Cuesta, J., Garcia-Sanchez, A., et al. (2013). Reducción de la densidad de población de jabalíes euroasiáticos (Sus scrofa) como medida para el control de la tuberculosis bovina: efectos en el jabalí y una población simparia de ciervos (Dama dama) en el centro de España. Anterior. Veterinario. Med. 110, 435-446. doi: 10.1016/j.prevetmed.2013.02.017
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Garnett, B. T., Delahay, R. J., y Roper, T. J. (2002). Uso de los recursos de la granja ganadera por parte de los tejones (Meles meles) y riesgo de transmisión de la tuberculosis bovina (Mycobacterium bovis) al ganado. Proc. Biol. Ciencia. 269, 1487–1491. doi: 10.1098/rspb.2002.2072
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Garnier, T., Eiglmeier, K., Camus, J.-C., Medina, N., Mansoor, H., Pryor, M., et al. (2003). La secuencia completa del genoma de Mycobacterium bovis. Proc. Natl. Acad. Ciencia. U. S. A. 100, 7877–7882. doi: 10.1073/pnas.1130426100
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Garrison, E., y Marth, G.. (2012). Detección de variantes basada en haplotipos de secuenciación de lectura corta.arXiv. [Epub antes de imprimir]. doi: 10.48550/arXiv.1207.3907
Ghebremariam, M. K., Rutten, V. P., Vernooij, J. C., Uqbazghi, K., Tesfaalem, T., Butsuamlak, T., et al. (2016). Prevalencia y factores de riesgo de tuberculosis bovina en el ganado lechero en Eritrea.Veterinario de BMC. Res. 12:80. doi: 10.1186/s12917-016-0705-9
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Glaser, L., Carstensen, M., Shaw, S., Robbe-Austerman, S., Wunschmann, A., Grear, D., et al. (2016). Epidemiología descriptiva y análisis de secuenciación del genoma completo para un brote de tuberculosis bovina en ganado vacuno y ciervos de cola blanca en el noroeste de Minnesota. PLoS One 11:e0145735. doi: 10.1371/journal.pone.0145735
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Goodchild, A. V., y Clifton-Hadley, R. S. (2001). Transmisión de ganado a ganado de Mycobacterium bovis. Tuberculosis 81, 23–41. doi: 10.1054/tube.2000.0256
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Gortazar, C., y Boadella, M. (2014). «C tuberculosis animal en España», en Zoonotic TuberculosisMycobacterium bovis y Other Pathogenic Mycobacteria. eds. C. O. Thoen, J. H. Steele y J. B. Kaneene (Estados Unidos: John Wiley & Sons).
Gortazar, C., Che Amat, A., y O’Brien, D. J. (2015). Preguntas abiertas y avances recientes en el control de una enfermedad infecciosa multihuésped: la tuberculosis animal. Mamífero Rev. 45, 160-175. doi: 10.1111/mam.12042
Gortazar, C., Torres, M. J., Vicente, J., Acevedo, P., Reglero, M., de la Fuente, J., et al. (2008). Tuberculosis bovina en la reserva de la biosfera de Donana: el papel de los ungulados salvajes como reservorios de enfermedades en los últimos bastiones de linces ibéricos. PLoS One 3:e2776. doi: 10.1371/journal.pone.0002776
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Verde, D. M., beso, yo. Z., Mitchell, A. P., y Kao, R. R. (2008). Estimaciones para la transmisión local y basada en el movimiento de la tuberculosis bovina en el ganado británico. Proc. Biol. Ciencia. 275, 1001–1005. doi: 10.1098/rspb.2007.1601
Guta, S., Casal, J., Garcia-Saenz, A., Saez, J. L., Pacios, A., Garcia, P., et al. (2014b). Factores de riesgo de persistencia de la tuberculosis bovina en manadas de carne de vacuno del sur y centro de España.Anterior. Veterinario. Med. 115, 173-180. doi: 10.1016/j.prevetmed.2014.04.007
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Guta, S., Casal, J., Napp, S., Saez, J. L., Garcia-Saenz, A., Perez de Val, B., et al. (2014a). Investigación epidemiológica de las desglosas de los retros de tuberculosis bovina en España 2009/2011. PLoS One 9:e104383. doi: 10.1371/journal.pone.0104383
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Hatherell, H. A., Colijn, C., Stagg, H. R., Jackson, C., Winter, J. R., y Abubakar, I. (2016). Interpretación de la secuenciación del genoma completo para investigar la transmisión de la tuberculosis: una revisión sistemática. BMC Med. 14:21. doi: 10.1186/s12916-016-0566-x
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Hauer, A., De Cruz, K., Cochard, T., Godreuil, S., Karoui, C., Henault, S., et al. (2015). Evolución genética de Mycobacterium bovis que causa tuberculosis en el ganado y la vida silvestre en Francia desde 1978.PLoS One 10:e0117103. doi: 10.1371/journal.pone.0117103
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Informe Final Técnico Tuberculosis Bovina Año (2020). Ministerio de Agricultura, Pesca, y Alimentación; 2021 Disponible en: https://www.mapa.gob.es/es/ganaderia/temas/sanidad-animal-higiene-ganadera/annexiiinformefinaltecnicotb2020_tcm30-564497.pdf (Consultado el 12 de septiembre de 2021).
Joshi, D., Harris, N. B., Waters, R., Thacker, T., Mathema, B., Krieswirth, B., et al. (2012). Los polimorfismos de un solo nucleótido en el genoma de Mycobacterium bovis resuelven las relaciones filogenéticas. J. Clin. Microbiol. 50, 3853–3861. doi: 10.1128/JCM.01499-12
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Kamerbeek, J., Schouls, L., Kolk, A., van Agterveld, M., van Soolingen, D., Kuijper, S., et al. (1997). Detección simultánea y diferenciación de cepas de Mycobacterium tuberculosis para diagnóstico y epidemiología. J. Clin. Microbiol. 35, 907–914. doi: 10.1128/jcm.35.4.907-914.1997
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Karolemeas, K., McKinley, T. J., Clifton-Hadley, R. S., Goodchild, A. V., Mitchell, A., Johnston, W. T., et al. (2011). Recurrencia de los descomposición de la tuberculosis bovina en Gran Bretaña: factores de riesgo y predicción. Anterior. Veterinario. Med. 102, 22-29. doi: 10.1016/j.prevetmed.2011.06.004
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Kato-Maeda, M., Ho, C., Passarelli, B., Banaei, N., Grinsdale, J., Flores, L., et al. (2013). Uso de la secuenciación del genoma completo para determinar la microevolución de Mycobacterium tuberculosis durante un brote. PLoS One 8:e58235. doi: 10.1371/journal.pone.0058235
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Lee, R. S., Radomski, N., Proulx, J. F., Manry, J., McIntosh, F., Desjardins, F., et al. (2015). Resurgimiento y amplificación de la tuberculosis en el Ártico canadiense. J. Infectar. Dis. 211, 1905–1914. doi: 10.1093/infdis/jiv011
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Li, H. y Durbin, R. (2009). Alineación de lectura corta rápida y precisa con diquejes-transformación de enredaje. Bioinformática 25, 1754–1760. doi: 10.1093/bioinformática/btp324
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Malone, K. M., Farrell, D., Stuber, T. P., Schubert, O. T., Aebersold, R., Robbe-Austerman, S., et al. (2017). Se ha actualizado la secuencia del genoma de referencia y la anotación de Mycobacterium bovis AF2122/97. Anotación del genoma 5, e00157–e00117. doi: 10.1128/genomeA.00157-17
Michelet, L., Conde, C., Branger, M., Cochard, T., Biet, F., y Boschiroli, M. L. (2019). Red de transmisión de la infección por Mycobacterium bovis transmitida por ciervos revelada por un enfoque WGS.Microorganismos 7:687. doi: 10.3390/microorganismos7120687
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Ministerio de Agricultura, Pesca y Alimentación (2021). Dirección General De Sanidad de la Producción Agraria: Ministerio de Agricultura, Pesca y Alimentación. Disponible en: https://www.mapa.gob.es/es/ganaderia/temas/sanidad-animal-higiene-ganadera/programatb2021versionabril_tcm30-561045.pdf (Consultado el 12 de septiembre de 2021).
Ministerio de Agricultura, Pesca, Alimentación y Medio Ambiente (2020). Plan de Actuación sobre TUBerculosis en Especies Silvestres (PATUBES). Disponible en: https://www.mapa.gob.es/es/ganaderia/temas/sanidad-animal-higiene-ganadera/patubes2017_3_tcm30-378321.pdf (Consultado el 5 de agosto de 2021).
Más, S. J., Radunz, B., y Glanville, R. J. (2015). Lecciones aprendidas durante la exitosa erradicación de la tuberculosis bovina de Australia. Veterinario. Rec. 177, 224–232. doi: 10.1136/vr.103163
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Munoz-Mendoza, M., Marreros, N., Boadella, M., Gortazar, C., Menendez, S., de Juan, L., et al. (2013). Tuberculosis de jabalí en la España atlántica ibérica: una imagen diferente de los hábitats mediterráneos. Veterinario de BMC. Res. 9:176. doi: 10.1186/1746-6148-9-176
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Naranjo, V., Gortazar, C., Vicente, J., y de la Fuente, J. (2008). Evidencia del papel del jabalí europeo como reservorio del complejo Mycobacterium tuberculosis. Veterinario. Microbiol. 127, 1–9. doi: 10.1016/j.vetmic.2007.10.002
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
O’Brien, D. J., Schmitt, S. M., Fitzgerald, S. D., Berry, D. E., y Hickling, G. J. (2006). Gestión del embalse de vida silvestre de Mycobacterium bovis: experiencia de Michigan, EE. UU. Veterinario. Microbiol. 112, 313-323. doi: 10.1016/j.vetmic.2005.11.014
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Orloski, K., Robbe-Austerman, S., Stuber, T., Hench, B., y Schoenbaum, M. (2018). Secuenciación del genoma completo de Mycobacterium bovis aislado del ganado en los Estados Unidos, 1989-2018.Delante. Veterinario. Ciencia. 5:253. doi: 10.3389/fvets.2018.00253
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Paradis, E., Claude, J. y Strimmer, K. (2004). APE: análisis de la filogenética y la evolución en el lenguaje R. Bioinformática 20, 289–290. doi: 10.1093/bioinformática/btg412
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Perea, C., Ciaravino, G., Stuber, T., Thacker, T. C., Robbe-Austerman, S., Allepuz, A., et al. (2021). El análisis del SNP del genoma completo identifica los grupos de transmisión de la supuesta Mycobacterium bovis en el ganado y la vida silvestre en Cataluña, España. Microorganismos. 9:1629. doi: 10.3390/microorganisms9081629
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Pérez-Lago, L., Navarro, Y., y García de Vila, D. (2014). Conocimiento actual y desafíos pendientes en la zoonosis causados por Mycobacterium bovis: una revisión. Res. Veterinario. Ciencia. 97, S94–S100. doi: 10.1016/j.rvsc.2013.11.008
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Pohlert, T. (2020). PMCMRplus: Calcule comparaciones múltiples por pares de sumas de rango media. Paquete R versión 1.4.4.
Pozo, P., Romero, B., Bezos, J., Grau, A., Nacar, J., Saez, J. L., et al. (2020). Evaluación de los factores de riesgo asociados con los rehados con una mayor duración de las descomposición de la tuberculosis bovina en Castilla y León, España (2010-2017). Delante. Veterinario. Ciencia. 7:545328. doi: 10.3389/fvets.2020.545328
Price-Carter, M., Brauning, R., de Lisle, G. W., Livingstone, P., Neill, M., Sinclair, J., et al. (2018). Secuenciación del genoma completo para determinar la fuente de las infecciones por Mycobacterium bovis en manadas de ganado y vida silvestre en Nueva Zelanda. Ciencia veterinaria frontal 5:272. doi: 10.3389/fvets.2018.00272
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Equipo central de R (2019). R: Un lenguaje y entorno para la computación estadística. Viena, Austria: Fundación R para la Computación Estadística.
Reis, A. C., Salvador, L. C. M., Robbe-Austerman, S., Tenreiro, R., Botelho, A., Albuquerque, T., et al. (2021). La secuenciación del genoma completo refina el conocimiento sobre la estructura de la población de Mycobacterium bovis a partir de un sistema de tuberculosis multiáfitrión.Microorganismos. 9:1585. doi: 10.3390/microorganisms9081585
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Reis, A. C., Tenreiro, R., Albuquerque, T., Botelho, A., y Cunha, M. V. (2020). La vigilancia molecular a largo plazo proporciona pistas sobre el origen del ganado de Mycobacterium bovis en Portugal.Ciencia. Representante. 10:20856. doi: 10.1038/s41598-020-77713-8
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Reviriego Gordejo, F. J., y Vermeersch, J. P. (2006). Hacia la erradicación de la tuberculosis bovina en la Unión Europea. Veterinario. Microbiol. 112, 101–109. doi: 10.1016/j.vetmic.2005.11.034
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Visor de genómica integradora. Nat. Biotechnol. 29, 24-26. doi: 10.1038/nbt.1754
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Rodriguez-Campos, S., Gonzalez, S., de Juan, L., Romero, B., Bezos, J., Casal, C., et al. (2012). Una base de datos para la tuberculosis animal (mycoDB.Es) en el contexto del programa nacional español para la erradicación de la tuberculosis bovina. Infectar. Genet. Evol. 12, 877-882. doi: 10.1016/j.meegid.2011.10.008
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Rodriguez-Campos, S., Romero, B., Bezos, J., de Juan, L., Alvarez, J., Castellanos, E., et al. (2010). Alta diversidad de spoligotype dentro de una población de Mycobacterium bovis: pistas para entender la demografía del patógeno en Europa. Veterinario. Microbiol. 141, 89-95. doi: 10.1016/j.vetmic.2009.08.007
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Roetzer, A., Diel, R., Kohl, T. A., Ruckert, C., Nubel, U., Blom, J., et al. (2013). Secuenciación del genoma completo frente al genotipado tradicional para la investigación de un brote de Mycobacterium tuberculosis: un estudio epidemiológico molecular longitudinal. PLoS Med. 10:e1001387. doi: 10.1371/journal.pmed.1001387
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Romero, B., Aranaz, A., Sandoval, A., Alvarez, J., de Juan, L., Bezos, J., et al. (2008). La persistencia y evolución molecular de la población de Mycobacterium bovis del ganado y la vida silvestre en el Parque Nacional Donana se revelan por la variación del genotipo. Veterinario. Microbiol. 132, 87-95. doi: 10.1016/j.vetmic.2008.04.032
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Salvador, L. C. M., O’Brien, D. J., Cosgrove, M. K., Stuber, T. P., Schooley, A. M., Crispell, J., et al. (2019). Manejo de enfermedades en la interfaz vida silvestre-ganado: uso de la secuenciación del genoma completo para estudiar el papel del alce en la transmisión de Mycobacterium bovis en Michigan, EE. UU. Mol. Ecol. 28, 2192–2205. doi: 10.1111/mec.15061
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Stamatakis, A. (2014). RAxML versión 8: una herramienta para el análisis filogenético y el postanálisis de grandes filogenias. Bioinformática 30, 1312–1313. doi: 10.1093/bioinformática/btu033
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Tonkin-Hill, G., Lees, J. A., Bentley, S. D., Frost, S. D. W., y Corander, J. (2018). RhierBAPS: Una implementación R del algoritmo de agrupación de población hierBAPS. Bienvenido, Abre Res. 3:93. doi: 10.12688/wellcomeopenres.14694.1
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Trewby, H., Wright, D., Breadon, E. L., Lycett, S. J., Mallon, T. R., McCormick, C., et al. (2016). Uso de la secuenciación bacteriana del genoma completo para investigar la persistencia y propagación local en la tuberculosis bovina. Epidemias 14, 26-35. doi: 10.1016/j.epidem.2015.08.003
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Wickham, H. (2009). ggplot2: Gráficos elegantes para el análisis de datos. Springer-Verlag Nueva York.
Wickham, H., François, R., Henry, L., y Müller, K.. (2019). Paquete R versión 0.4.3. R Encontrado. Estadísticas. Comput., Viena. Disponible en: https://CRAN.R-project.org/package=dplyr (Consultado el 12 de septiembre de 2021).
Palabras clave: Mycobacterium bovis, tuberculosis bovina, secuenciación del genoma completo, interfaz ganado-salvaje, vigilancia, filodinámica, transmisión entre especies
Cita: Pozo P, Lorente-Leal V, Robbe-Austerman S, Hicks J, Stuber T, Bezos J, de Juan L, Saez JL, Romero B y Alvarez J (2022) Uso de la secuenciación de genoma completo para desentrañar la diversidad genética de un estipligotipo de Mycobacterium bovis prevalecente en un escenario Delante. Microbiol. 13:915843. doi: 10.3389/fmicb.2022.915843
Editado por:
Caterina Guzmán-Verri, Universidad Nacional de Costa Rica, Costa Rica
Revisado por:
Lorraine Michelet, Agence Nationale de Sécurité Sanitaire de l’Alimentation, de l’Environnement et du Travail (ANSES), France
Maria Vitale, Instituto Experimental de Zooprofiláctico de Sicilia (IZSSi), Italia
Amandine Hauer, Universidad de Gdansk, Polonia
Flabio R. Araujo, Corporación Brasileña de Investigación Agrícola (EMBRAPA), Brasil
Copyright © 2022 Pozo, Lorente-Leal, Robbe-Austerman, Hicks, Stuber, Bezos, de Juan, Saez, Romero y Álvarez.
*Correspondencia: Pilar Pozo, ppozo@ucm.es
Descargo de responsabilidad: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o reclamación que pueda ser hecha por su fabricante no está garantizado o respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía