
Análisis de firma de selección basado en RNA-Seq para identificar huellas genómicas asociadas con el fenotipo de cola gorda en ovejas

Análisis de firma de selección basado en RNA-Seq para identificar huellas genómicas asociadas con el fenotipo de cola gorda en ovejas
 Hossein Abbasabadi1
Hossein Abbasabadi1 Mohammad Reza Bakhtiarizadeh1*
Mohammad Reza Bakhtiarizadeh1* Mohammad Hossein Moradi2
Mohammad Hossein Moradi2 John C. McEwan3
John C. McEwan3- 1Departamento de Ciencia Animal y Avícola, Colegio de Aburaihan, Universidad de Teherán, Teherán, Irán
- número arábigoDepartamento de Ciencias Animales, Facultad de Agricultura y Recursos Naturales, Universidad de Arak, Arak, Irán
- 3Centro Agrícola Invermay, AgResearch, Mosgiel, Nueva Zelanda
La comprensión de los antecedentes genéticos detrás del desarrollo de la cola gorda en las ovejas puede ser útil para desarrollar programas de mejoramiento genético, mientras que la base genética de la formación de la cola gorda aún no se comprende bien. Aquí, para identificar las regiones genómicas que influyen en el tamaño de la cola gorda en las ovejas, se realizó un análisis exhaustivo de identificación de firmas de selección mediante la comparación de razas de ovejas de cola gorda y cola fina. Además, para obtener los primeros conocimientos sobre el uso potencial de RNA-Seq para el análisis de identificación de firmas de selección, la llamada a SNP se realizó utilizando conjuntos de datos de RNA-Seq. En total, se analizaron 45 muestras de RNA-Seq de siete estudios de cohorte, y se utilizó el método FST para detectar las firmas de selección. Nuestros hallazgos indicaron que RNA-Seq podría ser de utilidad potencial para el análisis de identificación de firmas de selección. En total, se encontraron 877 SNPs relacionados con 103 genes en proceso de selección en 92 regiones genómicas. El análisis de anotación funcional reforzó la hipótesis de que los genes implicados en la oxidación de ácidos grasos pueden modular la acumulación de grasa en la cola de las ovejas y puso de manifiesto el posible papel regulador del proceso de angiogénesis en la deposición de grasa. De acuerdo con la mayoría de los estudios previos, nuestros resultados vuelven a enfatizar que el gen BMP2 es el objetivo de la selección durante la evolución de las ovejas. Un análisis adicional de la anotación génica de las regiones objetivo del proceso de evolución de las ovejas reveló que un gran número de genes incluidos en estas regiones están directamente asociados con el metabolismo de las grasas, incluidos los previamente reportados como candidatos involucrados en la morfología de la cola grasa de las ovejas, como NID2, IKBKG, RGMA, IGFBP7, UBR5, VEGFD y WLS. Además, una serie de genes, incluidos BDH2, ECHS1, AUH, ERBIN y CYP4V2, fueron de particular interés porque son genes bien conocidos asociados al metabolismo de las grasas y se consideran nuevos candidatos involucrados en el tamaño de la cola gorda. De acuerdo con el análisis de identificación de firmas de selección, el análisis de componentes principales agrupó las muestras en dos grupos completamente separados de acuerdo con las razas de cola gorda y delgada. Nuestros resultados proporcionan nuevos conocimientos sobre las bases genómicas de la diversidad fenotípica relacionada con la cola gorda de las razas ovinas y pueden utilizarse para determinar direcciones para mejorar las estrategias de cría en el futuro.
1 Introducción
La evidencia arqueológica sugiere que las ovejas fueron domesticadas por primera vez en el Medio Oriente hace aproximadamente 9.000-11.000 años (1). Desde entonces, las ovejas domésticas se han extendido por todo el mundo, ya que se han establecido más de 1.400 razas mediante la adaptación a una amplia gama de entornos en los que han sido criadas y criadas para diversos fines por los seres humanos (2). En general, las razas ovinas se pueden dividir en dos grupos principales, las de cola gorda (cola gorda corta, cola gorda larga y cola gorda rabadesca) y las de cola fina (colas largas y finas y colas cortas y finas), en términos de fenotipo de cola (3). Dado que las razas de cola gorda han sido criticadas por sus sistemas de producción intensivos, en términos de valor económico y costo energético, es necesario que los criadores de ovejas comprendan los mecanismos genéticos que controlan el desarrollo de la grasa de la cola para crear programas de cría destinados a reducir el tamaño de la cola (4). Sin embargo, la base genética de tales diferencias aún no se comprende bien. Se ha demostrado que las presiones de la selección natural y humana dejan firmas detectables en el genoma y pueden identificarse comparando razas con fenotipos extremos, como las razas de ovejas de cola gorda y fina (5). Por lo tanto, estas firmas de selección son cruciales para comprender cómo la domesticación y la cría de ovejas dieron forma a la estructura del genoma de las razas de cola gorda y fina. Esto se puede explorar a través de la investigación de las regiones genómicas de interés que potencialmente albergan barridos selectivos causados por la selección.
Un barrido selectivo es un proceso que ocurre cuando una variante genética particular o un alelo se vuelve rápidamente más común en una población debido a la selección natural o artificial (6). Las firmas de selección pueden detectarse examinando la distribución de las variaciones genéticas que rodean a un alelo seleccionado, lo que puede conducir a la identificación de una región con diversidad genética reducida en torno al alelo seleccionado (7). Se han hecho varios intentos de utilizar este enfoque en el contexto del desarrollo de la cola gorda (8-16). Por ejemplo, se llevaron a cabo dos experimentos independientes en estudios anteriores utilizando datos de genotipos de poblaciones de ovejas iraníes y el proyecto integral Sheep HapMap. Estos experimentos compararon razas de ovejas de cola fina y gorda, y el análisis confirmó asociaciones con la deposición de grasa en tres regiones genómicas en los cromosomas 5, 7 y X en ambos conjuntos de datos (10, 11). Además, Ahbara et al. (17) utilizaron el Illumina Ovine 50 K SNP BeadChip para identificar las relaciones genéticas y las regiones candidatas relacionadas con la deposición de grasa y la morfología de la cola relacionadas con 11 poblaciones de ovejas etíopes. Informaron de varios genes candidatos como NPR2, HINT2, SPAG8 e INSR asociados con la deposición de grasa, y ALX4, HOXB13 y BMP4 para la morfología de la cola. El análisis de la firma de selección de todo el genoma reveló ocho regiones candidatas que influyen en estos rasgos, proporcionando información sobre los mecanismos genéticos en las ovejas indígenas etíopes.
El genotipado por matrices se enfrenta a las tecnologías de secuenciación de alto rendimiento, la secuenciación de próxima generación (NGS). Por ejemplo, las matrices de SNP solo consideran una fracción de las variantes genéticas, una limitación que se vuelve más pronunciada cuando se aplica al análisis de razas locales. El rápido desarrollo de la NGS ha permitido identificar variantes genéticas de forma exhaustiva y eficiente, lo que es necesario para detectar firmas de selección genómica entre diferentes razas (18). Dado que la mayoría de los estudios anteriores utilizaron la matriz de SNP para genotipar animales, se han realizado pocas investigaciones para utilizar la secuenciación del genoma completo (WGS) para el escaneo genómico de barridos selectivos entre razas de ovejas de cola gorda y fina (19). Por el contrario, RNA-Seq se destaca como una de las aplicaciones de secuenciación más empleadas de NGS y se utiliza principalmente para analizar datos de transcriptoma. RNA-Seq captura información del genoma y se puede emplear para el descubrimiento de SNP. La viabilidad de descubrir variantes genéticas a partir de estos datos se demostró en nuestro estudio previo (4). Por lo tanto, los conjuntos de datos de RNA-Seq pueden considerarse una alternativa rentable a la WGS para la denominación de variantes genéticas (20). En este contexto, en los últimos 15 años se ha generado un gran número de conjuntos de datos de RNA-Seq que proporcionan un recurso aún no explotado de variantes genéticas en numerosas razas de ovejas de diferentes poblaciones. Por lo tanto, en el presente estudio, realizamos por primera vez un análisis de identificación de firmas de selección basado en los SNP identificados de siete estudios independientes de RNA-Seq (21-27). El objetivo principal de este estudio fue explorar el potencial del uso de conjuntos de datos de RNA-Seq para el análisis de identificación de firmas de selección. El objetivo secundario fue identificar regiones genómicas con fuertes evidencias de barridos selectivos a través de una comparación genómica entre razas de ovejas de cola gorda y delgada. Asumiendo que los barridos selectivos pueden resaltar genes clave asociados con la formación de cola de grasa, se realizó un análisis de enriquecimiento funcional en los genes ubicados en las regiones genómicas identificadas. Los conocimientos adquiridos en este estudio pueden proporcionar una base para futuras investigaciones sobre la mejora genética y la cría de razas ovinas.
2 Materiales y métodos
2.1 Conjunto de datos de RNA-Seq
Los conjuntos de datos de RNA-Seq se obtuvieron de estudios que se centraron en grupos de fenotipos extremos de razas ovinas en términos de tamaño de cola gorda. Por lo tanto, se realizó una revisión exhaustiva de la literatura y se encontró que siete estudios eran consistentes con nuestro propósito. En la Tabla 1 se proporciona una lista de estos conjuntos de datos. Todas las muestras se aislaron del tejido de la cola grasa de ovejas macho y se secuenciaron por pares en extremos, con un mínimo de tres repeticiones por raza. En total, se analizaron 45 muestras, incluidas 22 muestras de cola gorda y 23 muestras de cola fina, de nueve razas. Todos los datos se recuperaron de la base de datos GEO del NCBI y se analizaron sobre la base de la misma línea de bioinformática (Figura 1).
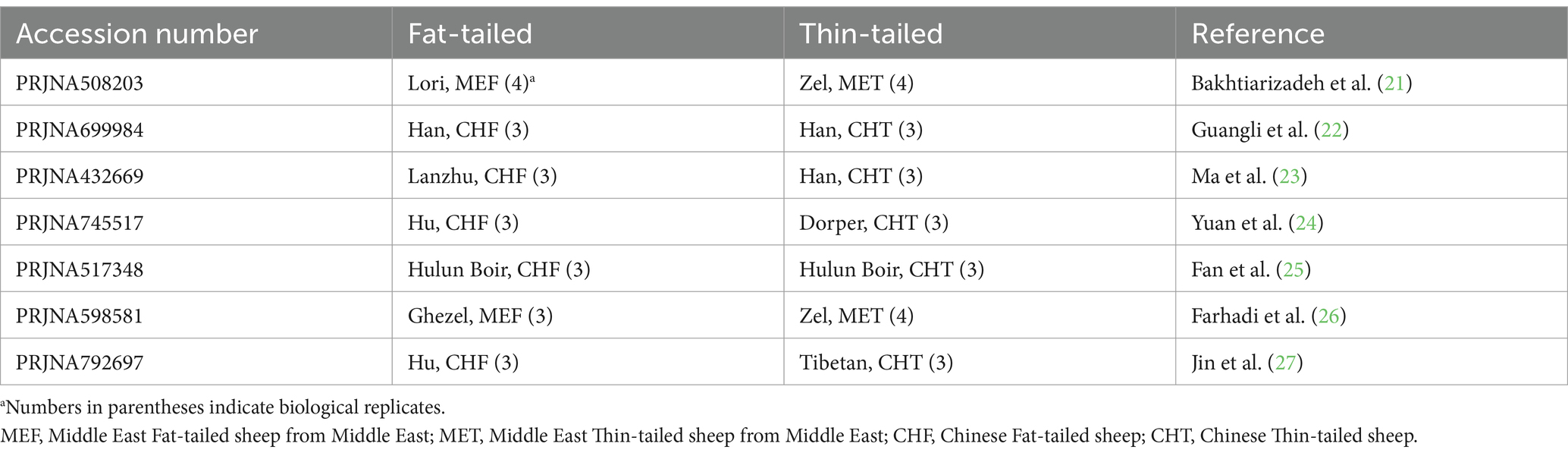
Tabla 1. Lista de conjuntos de datos utilizados en el presente estudio.
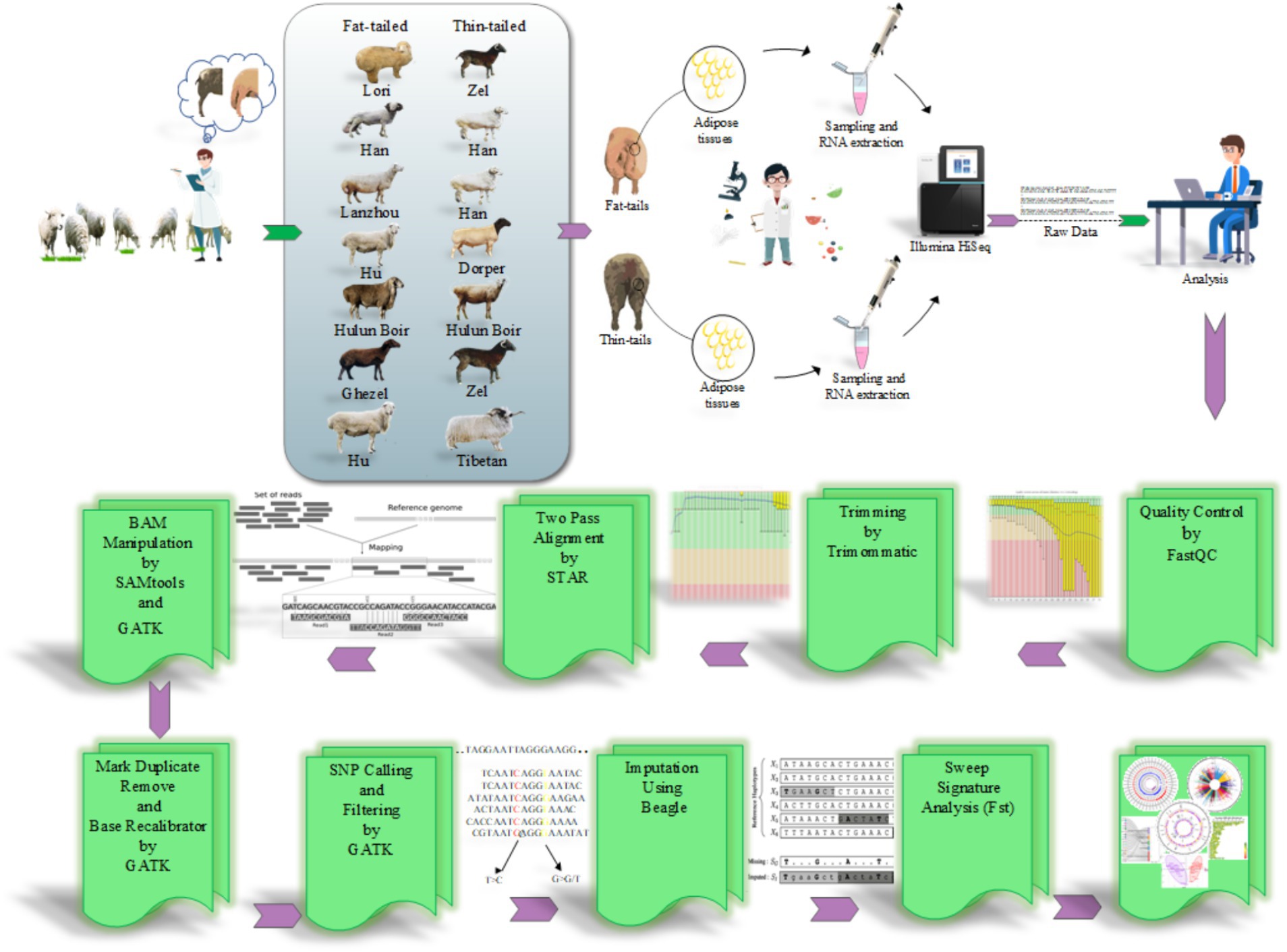
2.2 Control de calidad y mapeo
Para garantizar la calidad de los datos, los conjuntos de datos de RNA-Seq se sometieron inicialmente a un análisis de control de calidad utilizando el software FastQC (versión 0.11.5) (28). Además, se utilizó el software Trimmomatic (versión 0.38) para eliminar las lecturas/bases de baja calidad y las secuencias de adaptadores con el fin de mejorar la fiabilidad y la precisión de los datos al eliminar los artefactos no deseados y garantizar que solo se conservaran las lecturas de alta calidad para el análisis posterior (29). Los parámetros de recorte utilizados fueron los siguientes: TRAILING: 20, MAXINFO: 120:0.90 y MINLEN: 120 (21). Para mapear las lecturas limpias con el genoma de las ovejas (Rambouillet versión 1.0.104), se aplicó el software STAR (versión a2.7.9) de acuerdo con un enfoque de alineación de dos pasos (−-outFilterMismatchNoverLmax 0.06 y –outFilterMultimapNmax 10) (21). En este método, durante el proceso de alineación, se genera un nuevo archivo de índice que abarca uniones de empalme novedosas de alta confianza junto con sitios de unión de empalme conocidos. Por lo tanto, esta estrategia facilita la alineación con el genoma de referencia considerando tanto las uniones de empalme conocidas como las nuevas (30). La información conocida sobre el empalme se obtuvo del fichero GTF de ENSEMBL (Rambouillet v1.0.104, release 106) (31). Para minimizar los resultados falsos positivos, solo se conservaron las lecturas alineadas con los extremos emparejados de forma única y concordante para el análisis posterior. Posteriormente, las lecturas que se asignaron a la misma ubicación se marcaron utilizando la función MarkDuplicates de la herramienta GATK (versión 4.2.6.1) (32). Estas lecturas se consideraron duplicados de PCR y se excluyeron de análisis posteriores (33). Para aumentar la precisión de la llamada SNP, se realizó una recalibración de la puntuación de calidad base utilizando los módulos BaseRecalibrator y ApplyBQSR de la herramienta GATK (34) basada en la base de datos SNP ovina Ensembl (Ovis aries, Rambouillet).
2.3 Llamada y filtrado de SNP
Finalmente, se empleó el módulo HaplotypeCaller de GATK para llamar a las variantes putativas para cada muestra con un valor de stand_call_conf y stand_emit_conf de 30 y mbq de 25 (4). Las variantes iniciales identificadas se filtraron utilizando un conjunto universal de parámetros de filtrado, incluidos HomopolymerRun >5, Profundidad total de cobertura <10, QualitybyDepth <2, RMSMappingQuality <40, MappingQualityRankSum <-12.5 y ReadPosRankSum <-8 (4). Para mejorar la confiabilidad de los SNP identificados, se implementó un paso de filtrado adicional y se conservaron las variantes con al menos tres lecturas compatibles con el SNP. Además, se eliminaron las variantes ubicadas en regiones problemáticas, incluidas las regiones de baja complejidad (regiones de repeticiones de secuencia simple, ± 3 bases) o regiones con regiones de señal de transcripción bidireccional, exónicas y de empalme (dentro de 5 pb de la región de flanqueo intrónico) (4). Solo las variantes que cumplían estos criterios y se notificaron como SNP conocidos en la base de datos de SNP ovinos de Ensembl se conservaron para su posterior análisis. Este proceso de filtrado tenía como objetivo mejorar la calidad y la fiabilidad de los SNP identificados para el análisis posterior.
2.4 Imputación
Para mejorar la completitud de la información genotípica, se implementó la imputación de genotipos. El archivo SNP de referencia relacionado con el genoma de referencia ovino se obtuvo de la base de datos Animal-ImputeDB. El panel de referencia contenía 29.889.815 SNP de 450 muestras relacionadas con 43 razas de ovejas domésticas, lo que garantizaba un fondo genético diverso y proporcionaba un conjunto de datos de alta resolución (35). Dado que las coordenadas genómicas de los SNPs en esta base de datos se basaban en un genoma de oveja diferente (Oar_v3.1), se utilizó la herramienta LiftOver (versión 46e) (36) para convertir las coordenadas genómicas de Oar_v3.1 a Rambouillet v1.0.104. Para ello, todos los SNPs se convirtieron a las coordenadas Rambouillet v1.0.104 con el archivo de cadena correspondiente obtenido del UCSC Genome Browser. Posteriormente, se utilizó el software Beagle (versión 5.4) (37, 38) basado en el modelo de frecuencia de haplotipos para imputar los SNPs. Este modelo supone que no falta ningún genotipo en los genotipos de referencia para simplificar los cálculos. La imputación de genotipos se basa en la identidad por descendencia (EII), donde dos partes del cromosoma se heredan de un ancestro común sin recombinación ni cruzamiento. Al identificar con precisión los segmentos de EII, los alelos no tipificados en el haplotipo objetivo se pueden copiar del haplotipo de referencia, lo que aumenta efectivamente el número de marcadores (39, 40). Se mantuvieron SNPs con una probabilidad de predicción superior al 90% (según el valor de DR2, correlación cuadrada entre los genotipos imputados y reales) y un MAF > 0,05 (frecuencia de alelos raros o menos comunes) para optimizar la precisión de la imputación para análisis posteriores (41). Para evaluar la precisión de los genotipos imputados, se utilizó un análisis de concordancia. Para ello, se seleccionó aleatoriamente y enmascaró el 1% de los SNPs comunes entre los SNPs identificados y el fichero de referencia (de Animal-ImputeDB) antes de la imputación. El resto de los SNPs se imputaron y se aplicaron para comprobar si los SNPs enmascarados se habían predicho correctamente o no (42). Este proceso se repitió cinco veces para garantizar la fiabilidad de los resultados.
2.5 Análisis de identificación de firma de selección
Para detectar signos de selección entre los dos grupos distintivos, se estimaron dos métodos complementarios, el índice de fijación (FST, propuesto por Wright) (43) y el índice de fijación insesgado (Theta, propuesto por Weir y Cockerham) (44). Para ello, se agruparon las razas de cola gorda y cola fina, y luego se calcularon los estadísticos FST y Theta para estos dos grupos (grupo de cola gorda: 22 muestras y grupo de cola fina: 23 muestras). Estos índices de diferenciación poblacional cuantifican el nivel de diferenciación genética entre las razas ovinas de cola gorda y cola fina en función de las diferencias en las frecuencias alélicas. Los valores de los índices de fijación pueden oscilar teóricamente entre 0 (que no muestra diferenciación) y 1 (que indica una diferenciación completa, es decir, las poblaciones están fijas para diferentes alelos). Las regiones con valores extremos de F,ST y Theta pueden considerarse como buenas candidatas para barridos selectivos. En el presente estudio, se seleccionaron todas las ventanas extremas en los percentiles 99.9 (0.1% superior de FST y Theta) como portadoras de las regiones genómicas con firmas de selección. Se eligió una ventana de 40 kb porque parecía proporcionar una mejor señal que otros tamaños de ventana arbitrarios (45). Además, se exploró la relación genética entre las razas ovinas de cola gorda y cola fina mediante el análisis de componentes principales (PCA) basado en los SNPs localizados en las regiones genómicas identificadas como bajo selección positiva en razas de cola fina y gorda. Este análisis tuvo como objetivo determinar cómo los animales se asignaron a los grupos utilizando estos marcadores de baja densidad seleccionados positivamente. Todos los scripts para estimar los índices de Wright (FST) y Weir y Cockerham (Theta), así como el PCA, se escribieron y realizaron en R v 4.0.2.
2.6 Análisis funcional
Las regiones candidatas identificadas se anotaron utilizando la herramienta SNPeff (versión 5.1) (46) para detectar los genes que albergaban los SNPs en estas regiones de acuerdo con un archivo de anotación del genoma de referencia (ENSEMBL GTF, Rambouillet v1.0.104, release 106). Para obtener información sobre las funciones potenciales de estos genes, se realizó un análisis de enriquecimiento funcional utilizando la herramienta EnrichR. Esta herramienta es un completo y popular motor de búsqueda de servidor web de análisis de enriquecimiento de conjuntos genéticos que permite a los investigadores analizar y visualizar listas de genes para términos enriquecidos a través de la conexión a otras bases de datos importantes como GO, KEGG, etc. (47). Los procesos biológicos con un FDR <0,05 se consideraron términos significativos.
3 Resultados
3.1 Análisis del conjunto de datos RNA-Seq
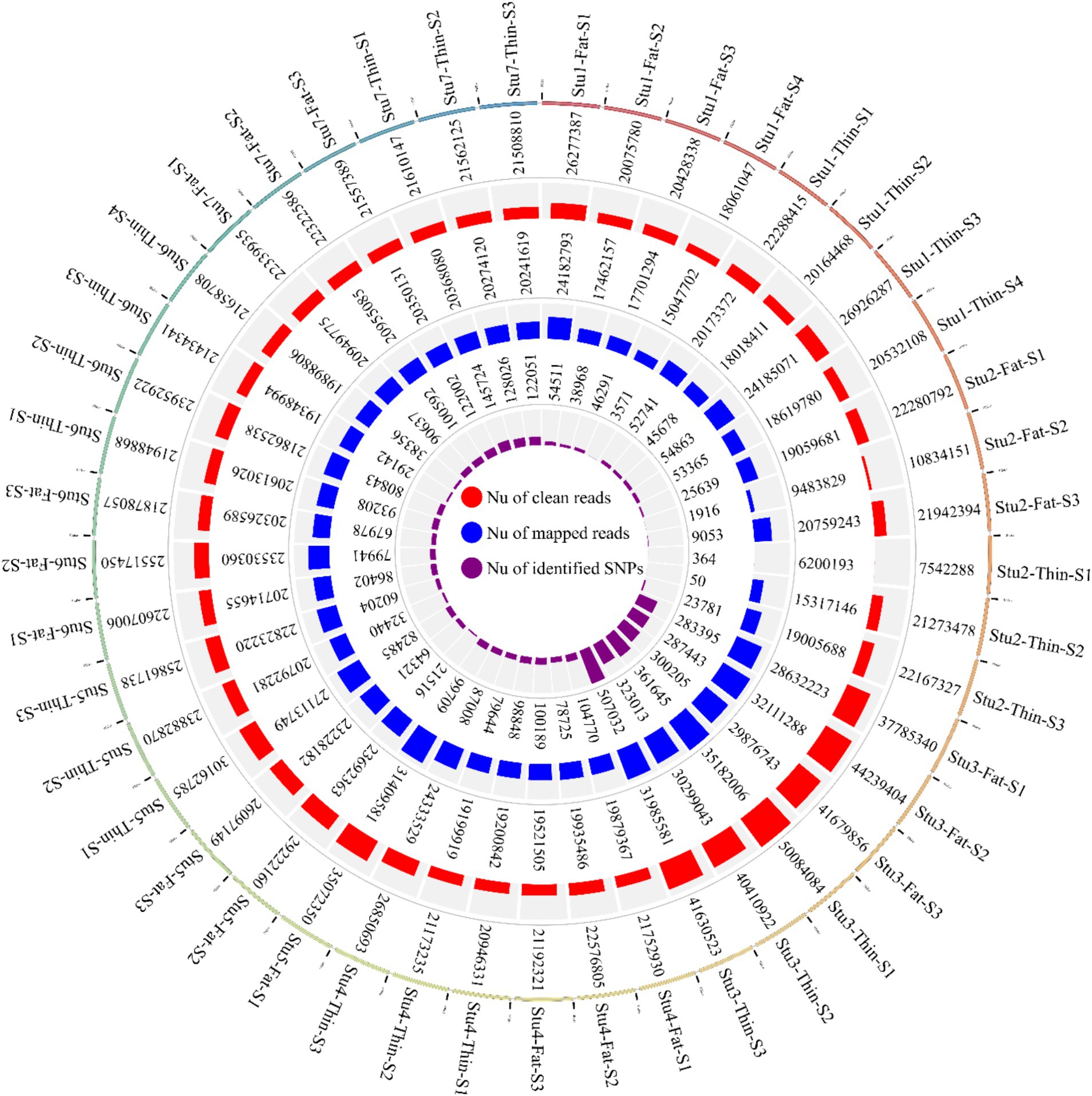
En este estudio, se analizaron siete conjuntos de datos de RNA-Seq relacionados con 22 ovejas de cola gorda y 23 ovejas de cola fina. El número total de lecturas sin procesar obtenidas para los siete conjuntos de datos fue de 1.136.651.699; con una media de 25 millones de lecturas por muestra. De estas, 556.807.475 y 579.844.224 lecturas pertenecían a las razas de cola gorda y fina, respectivamente. Tras el control de calidad y el filtrado, quedaron 1.131.314.100 lecturas (Figura 2). A continuación, las lecturas limpias se alinearon con el genoma de referencia de las ovejas, y 973.867.046 lecturas se alinearon con éxito con el genoma, con una tasa de alineación general del 93% (Figura 2). Las tasas generales de alineamiento entre los dos grupos fueron similares, con un 92,6% ± 0,05 para las razas de cola gorda y un 92,5% ± 0,06 para las razas de cola fina. De las lecturas alineadas, 973.867.046 lecturas se asignaron de forma única al genoma y se conservaron para su posterior análisis. La tasa media de alineación de las lecturas asignadas de forma única por muestra fue del 92,6 %, oscilando entre el 77,1 y el 98,5 % (material complementario S1).
3.2 Llamadas SNP
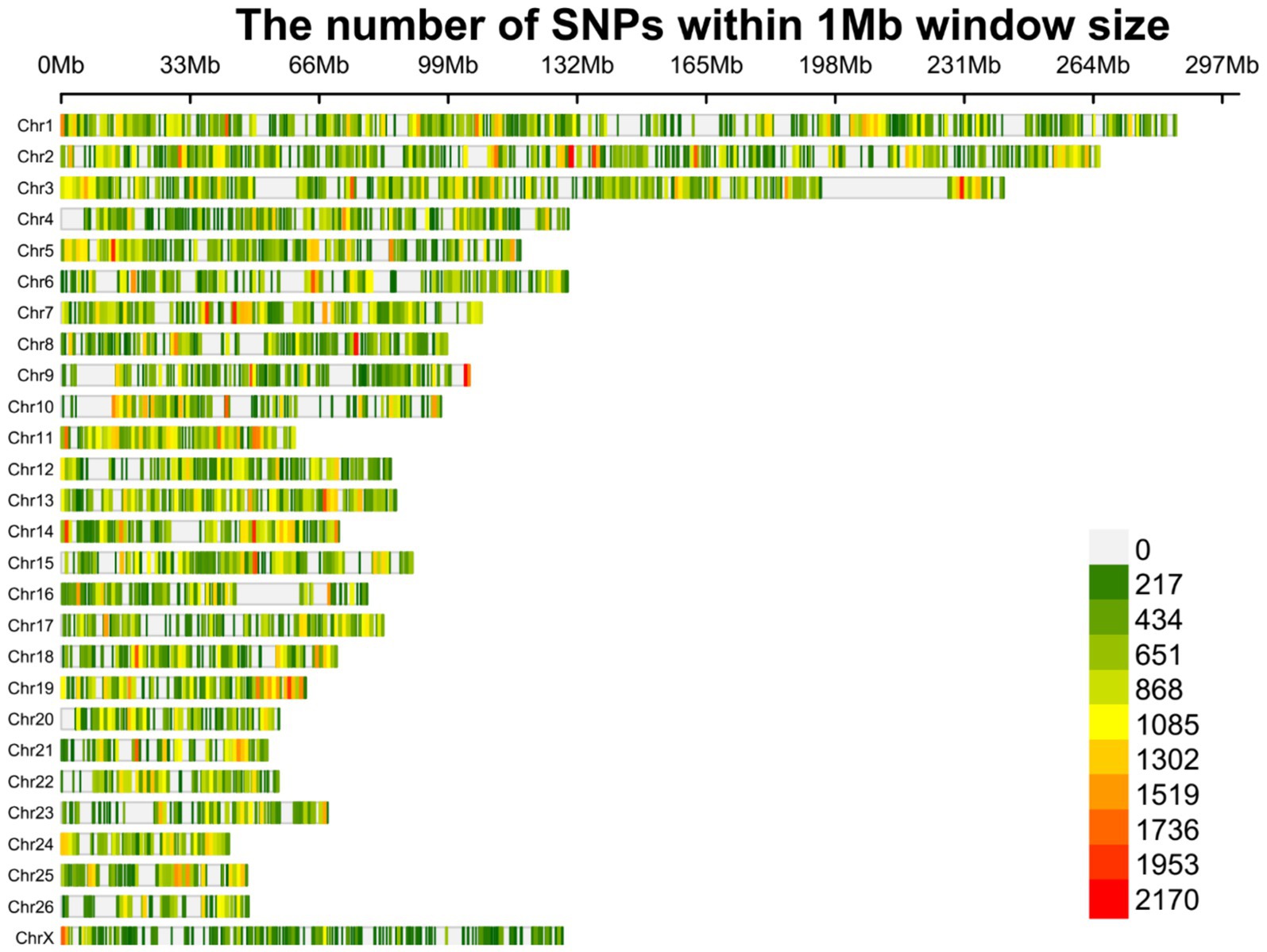
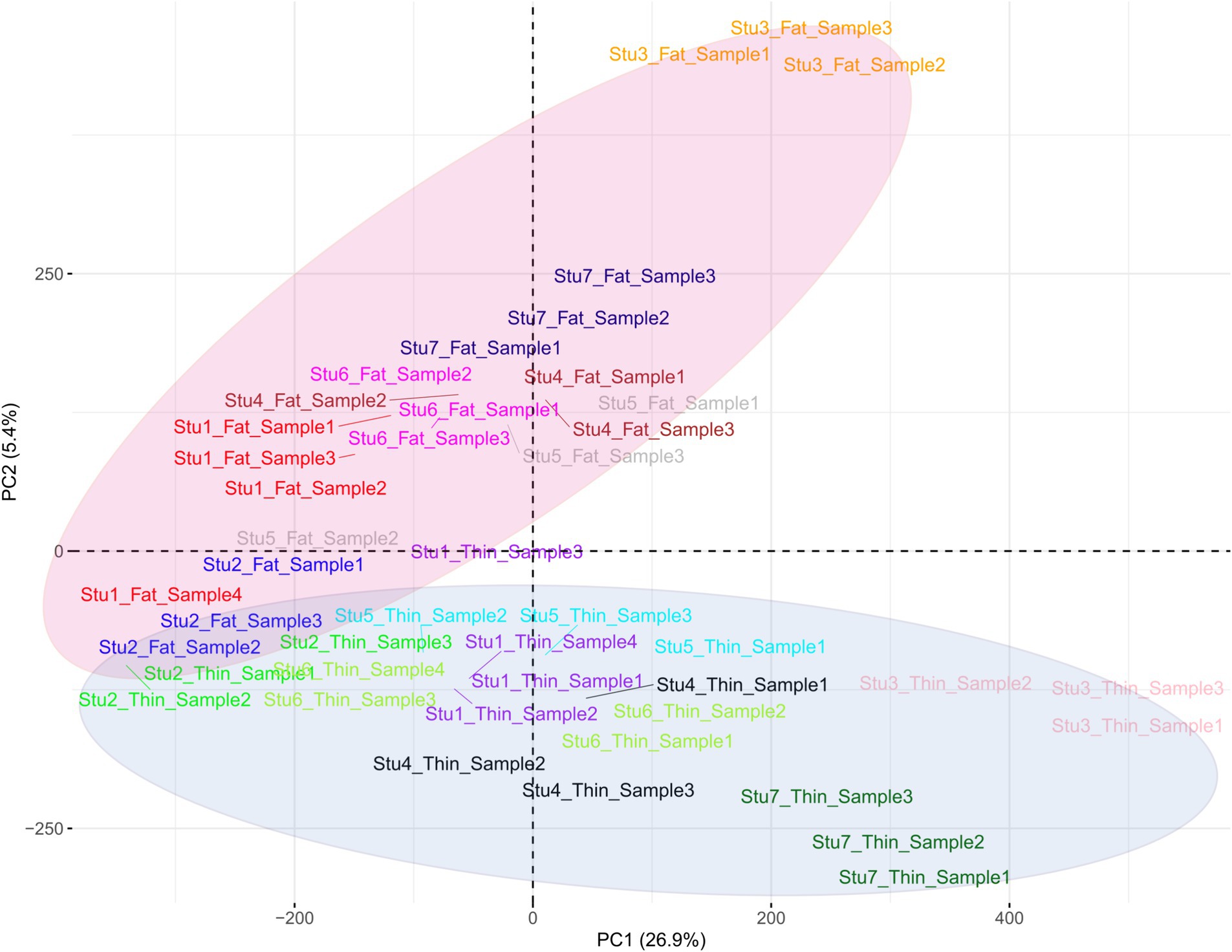
En total, se identificaron 1.719.262 variantes bialélicas únicas en todas las muestras en función de nuestra estricta cartera de productos. Entre estas, 1.415.762 variantes se informaron como SNP conocidos después de la comparación con la base de datos de dbSNP de ovejas. Vale la pena destacar que solo se aplicaron los SNPs reportados en la base de datos dbSNP de ENSEMBL para aumentar la confiabilidad de los resultados. La lista completa de los SNPs identificados por muestra se proporciona en el material complementario S2. Posteriormente, se imputaron estos SNPs (y se filtraron con DR2 > 0,90 y MAF > 0,05), y se encontraron 18.577.286 SNPs en todas las muestras, incluyendo 1.171.918 SNPs únicos. De todos los SNP, se detectaron 8.981.485 SNP en razas de cola gorda, mientras que se identificaron 9.595.801 SNP en razas de ovejas de cola fina. El número medio de SNPs detectados por muestra en las razas ovinas de cola gorda y cola fina fue de 408.249 y 417.208, respectivamente. El anillo interior del diagrama Circos de la Figura 2 representa el número de SNPs identificados por muestra de cada estudio. Entre los SNP únicos, el mayor número de SNP se encontró en el cromosoma 1 (128.394), y los números más bajos se localizaron en el cromosoma 26 (16.153). Además, se encontraron 24.977 SNPs en el cromosoma X. La Figura 3 muestra la densidad de estos SNPs por cromosoma. El análisis de concordancia del análisis de imputación reveló que los genotipos imputados fueron altamente consistentes en base a las cinco repeticiones, con una tasa de concordancia del 99%. Esto indica que los genotipos imputados se recuperaron con una precisión muy alta. Para investigar la estructura poblacional, se aplicaron todos los SNPs identificados para realizar análisis de PCA. El primer y segundo componente principal (PC1 y PC2) del PCA diferenciaron las razas de cola gorda y fina (Figura 4). En general, los dos primeros CP explicaron el 32,3% (PC1 = 26,93%, PC2 = 5,4%) de la variación en todos los datos genómicos.
3.3 Análisis de identificación de firma de selección
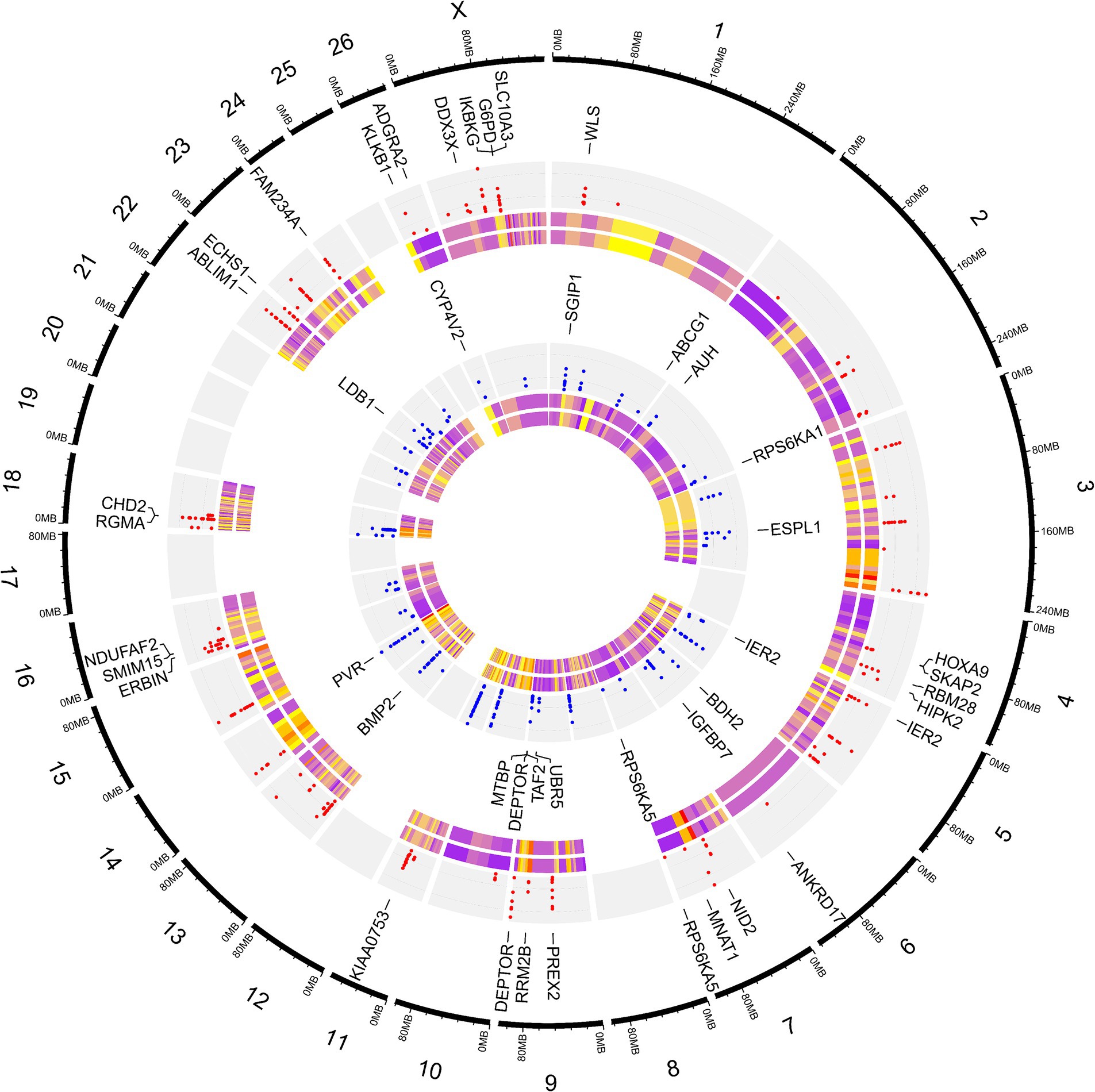
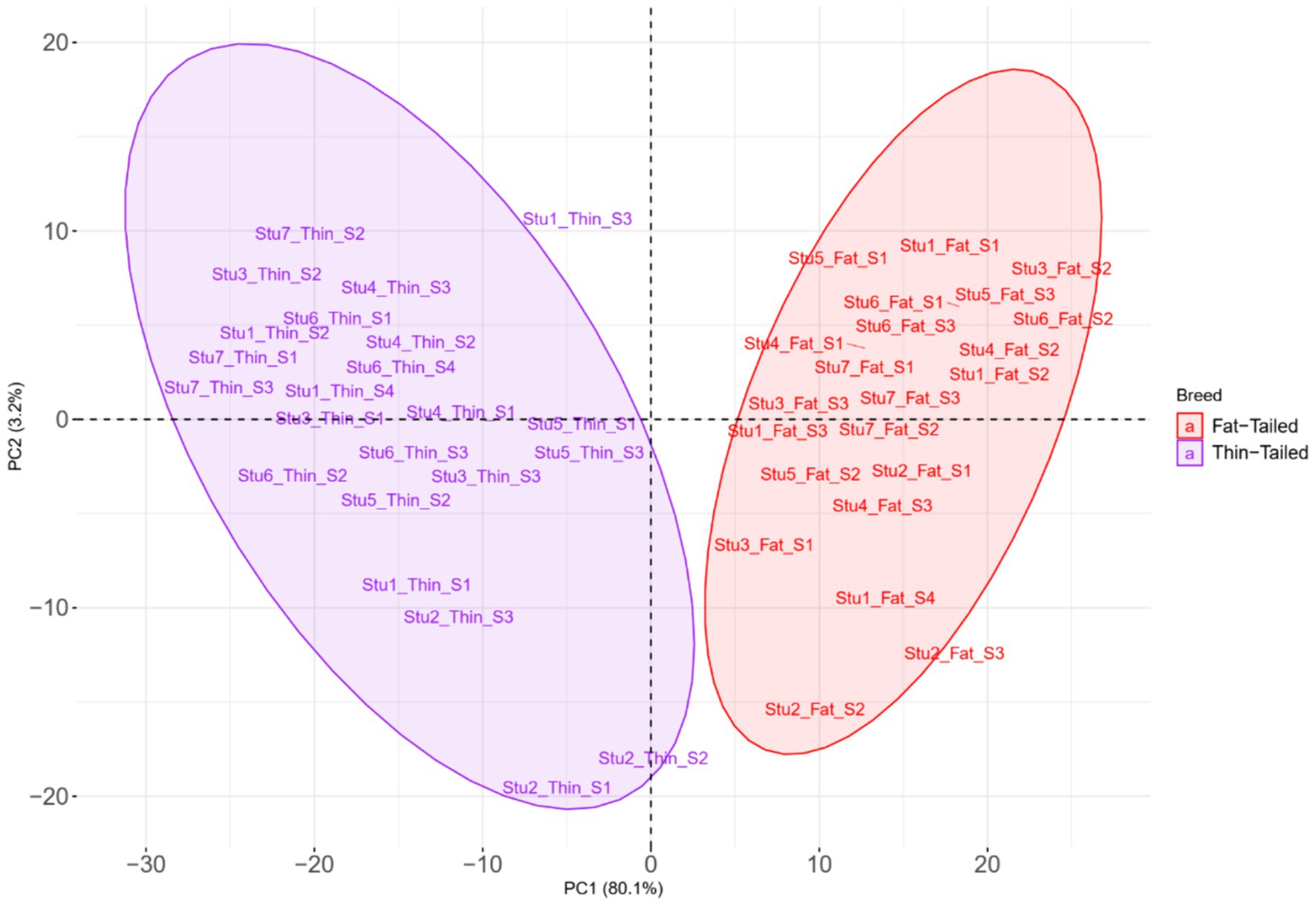
Para dilucidar las diferencias debidas a la selección e identificar las regiones seleccionadas y los genes candidatos involucrados en el desarrollo de la cola gorda, se estimaron los valores de FST por pares en todo el genoma entre las razas de cola gorda y cola fina sobre la base de 1.171.918 genotipos SNP (Figura 5). La distancia media entre los SNPs adyacentes en todo el genoma fue de 2,35 kb, lo que proporciona una alta resolución para detectar con precisión las firmas de selección. Para evaluar el grado de diferenciación de las razas debido a la estructura genética, se estimaron los estadísticos FST y Theta, que evalúan la proporción de la varianza genética dentro y entre las subpoblaciones. Si bien el método FST de Wright no considera el error de muestreo, Weir y Cockerham propusieron un estimador FST insesgado (Theta) que tiene en cuenta el tamaño de la muestra (11). El coeficiente de correlación de Pearson entre los dos métodos en nuestro estudio reveló una correlación positiva fuerte y significativa (r2 = 0,97); por lo tanto, solo se utilizó el FST para un análisis posterior (material complementario S3). El promedio de FST en todo el genoma fue de 0,015 (SD = 0,020). Se identificaron evidencias de selección para 877 SNPs dentro de 92 regiones, que superaron el punto de corte (valores extremadamente altos en el 0,1% de la cola derecha de la distribución FST) y se consideraron regiones del genoma que exhibieron una diferenciación poblacional significativa. En otras palabras, estas regiones podrían ser candidatas potenciales bajo selección positiva. Los valores de FST de estos SNPs oscilaron entre 0,15 y 0,28, con un promedio de 0,18. Estos SNPs se distribuyeron en todos los cromosomas (excepto 12, 17, 19 y 25), ya que el mayor y el menor número de SNPs se localizaron en los cromosomas 18 (86) y 21 (3), respectivamente. Además, se identificaron 103 genes, incluidos 87 genes con anotaciones de símbolos genéticos, en estas regiones; Estos genes pueden ser considerados como genes seleccionados positivamente involucrados en el desarrollo de la cola gorda en las ovejas. La distribución de estas firmas en todo el genoma se muestra en la Figura 6 como un gráfico de Circos. Además, se puede encontrar información más detallada sobre estas regiones genómicas en el material complementario S4. Utilizando PCA y en base a los dos primeros componentes principales (PC1 vs. PC2), las muestras se dividieron en dos grupos (cola gorda y cola fina), de acuerdo con los SNPs encontrados en las regiones de firma de selección. Los porcentajes de varianza explicados por PC1 y PC2 fueron de 80,1 y 3,2%, respectivamente (Figura 7).
3.4 Análisis de enriquecimiento funcional
La interpretación funcional de los genes identificados en las regiones seleccionadas reveló un enriquecimiento de 129 procesos biológicos con valores de p <0,05. Como se enumera en el material suplementario S5, algunos de los términos estaban relacionados con el metabolismo de los ácidos grasos, como «Beta-oxidación de ácidos grasos», «Oxidación de ácidos grasos» y «Proceso catabólico de ácidos grasos». De estos, cinco términos fueron significativos después de la corrección de múltiples pruebas por el método FDR, incluyendo «Angiogénesis germinada», «Oxidación de ácidos grasos», «Regulación positiva de la transición de fase del ciclo celular mitótico», «Vía de señalización del receptor de la superficie celular involucrada en la señalización célula-célula» y «Regulación positiva de la transición G1/S del ciclo celular mitótico» (Figura 8). Además, se anotaron directamente 10 genes asociados con procesos relacionados con el metabolismo de las grasas, incluidos BMP2, BDH2, ECHS1, ABCG1, AUH, CYP4V2, DDX3X, ERBIN, SLC10A3, WLS e IKBKG, que están marcados en la Figura 8.
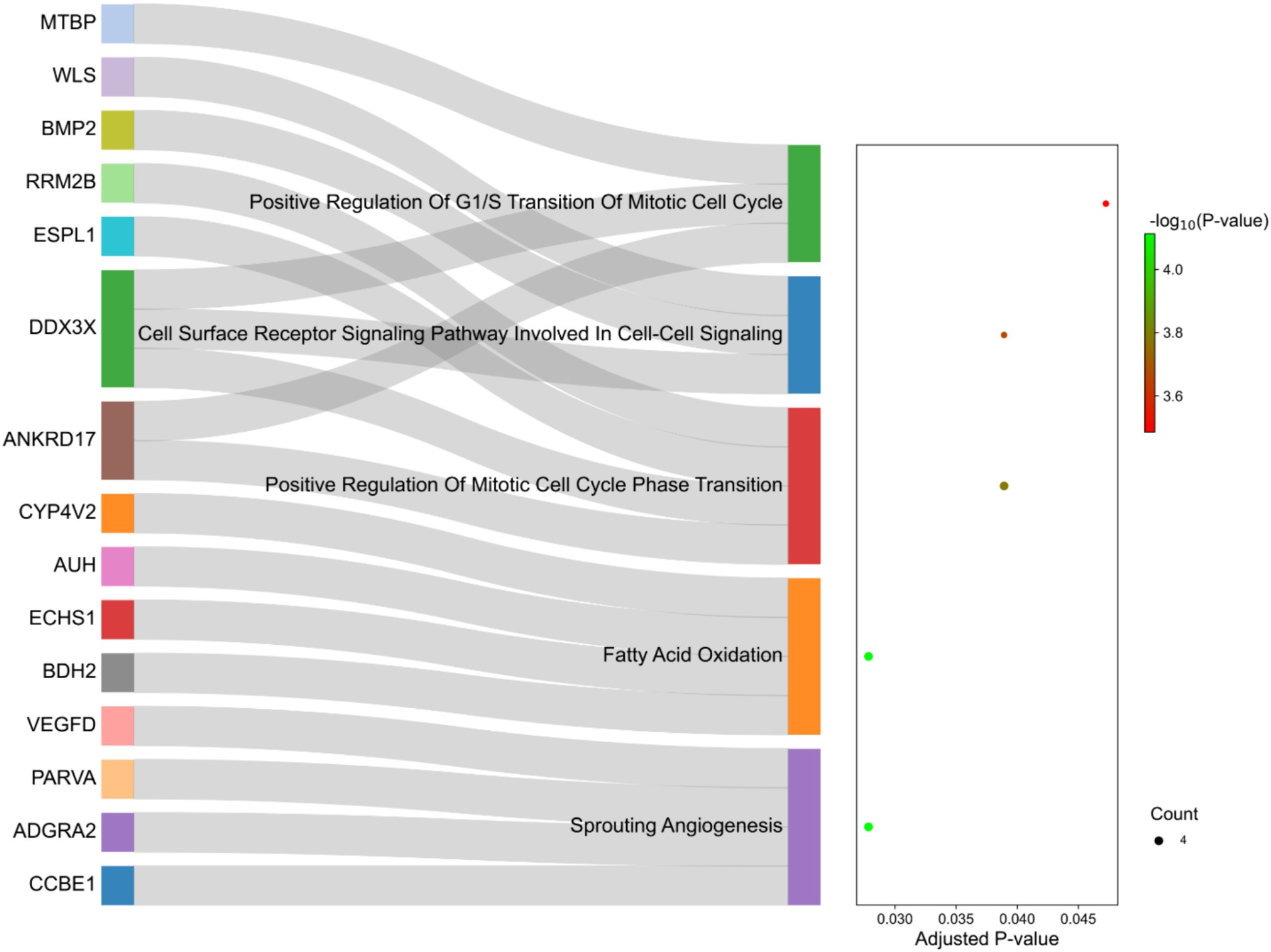
4 Discusión
Los animales domésticos han sido sometidos a selección natural y artificial, que les ha dejado características genómicas distintivas. Una de estas características es la reducción de la diversidad genética en las regiones a las que se dirigen rasgos específicos, como la forma de la cola gorda. En este sentido, el escaneo de todo el genoma para las firmas de selección es un método poderoso para identificar regiones candidatas que están sujetas a la diversidad fenotípica de las razas ovinas. A pesar de los numerosos estudios dedicados a identificar las firmas de selección en las ovejas, la base genética de varias formas de cola gorda en diferentes razas de ovejas no se ha dilucidado completamente. Para abordar esta brecha, es fundamental discernir la variación genómica entre las razas de ovejas de cola gorda y delgada. La evidencia de las firmas de selección ofrece información sobre los posibles factores genéticos relacionados con la deposición de grasa. Esta diferencia proporciona una oportunidad ideal para dilucidar los mecanismos moleculares que subyacen al almacenamiento de energía y al metabolismo de los lípidos, ya que las ovejas pueden ser modelos animales útiles para la investigación biomédica. Aprovechando los datos de RNA-Seq, el objetivo del presente estudio fue utilizar un análisis genómico comparativo entre razas de ovejas de cola gorda y cola fina para identificar firmas putativas de selección que pueden estar relacionadas con la formación de cola gorda. Para ello, se utilizaron siete razas ovinas de cola gorda y cinco de cola fina (Tabla 1). En particular, estas razas se pueden utilizar para identificar las firmas de selección general involucradas en la deposición de grasa en las colas de ovejas basadas en la hipótesis de que la fisiología de los órganos se conserva en todos los animales. En este contexto, hay varias vías que están profundamente conservadas entre las especies (48, 49). Según esta hipótesis, los roedores u otros animales pueden utilizarse como modelos para estudiar enfermedades humanas o procesos biológicos. Por lo tanto, las señales genómicas relacionadas con la deposición de grasa se conservan entre diferentes razas de ovejas y se pueden inferir comparando razas de ovejas de cola gorda y cola fina. Las firmas de selección denotan las regiones genómicas influenciadas por la selección positiva, lo que resulta en una mayor frecuencia de alelos beneficiosos dentro de una población.
Se han desarrollado varios métodos para detectar estas firmas, incluidos los enfoques basados en frecuencias alélicas como F, ST (método de Wright) y Theta (método de Weir y Cockerham) (11), y métodos basados en haplotipos como la puntuación de homocigosidad de haplotipos y hapFLK. Se demuestra que los procedimientos basados en haplotipos tienen el mayor poder para detectar la selección en curso, ya que exploran la estructura de los haplotipos y, esencialmente, identifican haplotipos inusualmente largos que llevan los alelos ancestrales y derivados (50). Sin embargo, el fenotipo de la cola de oveja es el resultado de una selección completa, no una selección continua (51). Por otro lado, los métodos basados en haplotipos necesitan SNPs de alta densidad y fases para el análisis de haplotipos (50), que no están garantizados por los datos de RNA-Seq. Por lo tanto, en el presente estudio, se aplicaron dos métodos de frecuencias alélicas que mostraron alta concordancia. El método FST es especialmente adecuado para detectar regiones que han estado bajo selección durante mucho tiempo y que se han acumulado en diferentes poblaciones. Este método capta los patrones históricos de variación y divergencia que han ocurrido a lo largo de muchas generaciones (52). En este sentido, en el presente estudio se aplicó el método FST, teniendo en cuenta desde la perspectiva de la genética de poblaciones que las razas de cola gorda evolucionaron a partir de su ancestro de cola fina hace aproximadamente 5.000 años (51). Vale la pena señalar que el número de SNPs identificados en las razas de cola fina (2.498.980) fue mayor que el de las razas de cola gorda (2.066.424), lo que apoya aún más esta hipótesis (Material suplementario S2).
Más del 82% de las variantes identificadas en nuestro análisis primario de llamadas de SNP se encontraron en la dbSNP (base de datos de SNP ovino Ensembl), lo que enfatiza la alta calidad y confiabilidad de nuestros datos de genotipo de SNP. Sin embargo, ~18% de los SNPs identificados no estaban disponibles en la base de datos dbSNP, lo que demuestra que es necesario hacer más intentos para perfilar la diversidad genética de las ovejas. En comparación con estudios anteriores que utilizaron datos de secuenciación del genoma completo, identificamos un número significativamente menor de SNP. Este hallazgo está completamente en línea con este punto: la mayoría de los SNP detectados por los datos de secuenciación del genoma completo se encuentran en regiones intergénicas o intrónicas (53-55). En este caso, para eliminar las variantes resultantes de los sitios de edición de ARN y maximizar la fiabilidad de los resultados, solo se consideraron los SNP conocidos para un análisis posterior (56, 57). En este estudio, se detectó un número relativamente grande de señales de selección (92 regiones genómicas, material suplementario S4), lo que refleja la complejidad del fenotipo de cola gorda. Sin embargo, algunas de estas señales pueden estar relacionadas con otros rasgos y se necesitan más investigaciones. El mayor número de firmas de selección se encontró en los cromosomas 18, X y 11 (material suplementario S4), lo que sugiere una mayor presión de selección sobre estos cromosomas.
Se realizó una anotación funcional de las regiones de firma de selección identificadas para investigar su papel específico en la deposición de grasa en la cola de las ovejas. Los dos términos más significativos fueron «Oxidación de ácidos grasos» y «Angiogénesis germinante». Curiosamente, y en total acuerdo con nuestros estudios previos (4, 21), los procesos biológicos asociados a la oxidación de ácidos grasos se enriquecieron significativamente en los genes seleccionados. Algunos genes importantes relacionados con estos procesos biológicos fueron BDH2, ECHS1, AUH y CYP4V2 (material suplementario S5). BDH2, 3-hidroxibutirato deshidrogenasa 2, está involucrada en la cetogénesis (síntesis y degradación de cuerpos cetónicos) y puede estimular el almacenamiento de grasa y la adiposidad al proporcionar precursores para la síntesis de lípidos y esteroles (58). Por otro lado, el papel de este gen en la oxidación de ácidos grasos ha sido destacado en estudios previos (59, 60). El segundo paso de la vía de β-oxidación es catalizado por ECHS1. En este sentido, se ha informado que la regulación positiva de este gen se asocia con una reducción en la deposición de grasa abdominal en pollos de engorde a través de la promoción de la oxidación de ácidos grasos (61). Se demostró que la expresión de ECHS1 en el tejido hepático de ratones alimentados con una dieta alta en grasas disminuyó y condujo a una producción acelerada de acetil coenzima, que es el precursor de la síntesis de ácidos grasos (62). Estos hallazgos proporcionan un apoyo adicional a nuestra hipótesis anterior, que sugiere que los genes involucrados en la oxidación de ácidos grasos pueden promover este proceso biológico y modular la acumulación de grasa en la cola de las razas ovinas. Teniendo en cuenta que las razas de cola gorda se originan a partir de ovejas de cola fina, se puede plantear la hipótesis de que las duras condiciones ambientales pueden haber llevado a la selección tanto artificial como natural, favoreciendo los alelos que regulan a la baja los genes involucrados en la oxidación de ácidos grasos en la cola de las ovejas. Vale la pena señalar que ninguno de los genes discutidos se informó previamente como genes candidatos involucrados en el desarrollo de la cola gorda en las ovejas y pueden considerarse genes nuevos asociados con este rasgo. En cuanto al proceso de «angiogénesis germinal», está bien documentado que el desarrollo de obesidad se asocia con adipogénesis y angiogénesis (63-65). El proceso de formación de nuevos vasos sanguíneos, la angiogénesis, se reporta como un paso limitante de la tasa en la expansión del tejido graso. En este sentido, la vasculatura es responsable del transporte de ácidos grasos a otros tejidos, así como del suministro de oxígeno y nutrientes, que son necesarios para la expansión del tejido graso (64). Los miembros de la familia de los VEGF (factores de crecimiento endotelial vascular) se consideran los principales componentes angiogénicos en el tejido adiposo y mostraron una alta expresión en los adipocitos y el tejido adiposo de ratones obesos (66). Un miembro importante de esta familia es el VEGFD (detectado en las regiones candidatas en el presente estudio), que desempeña un papel crucial en el desarrollo del tejido adiposo (65). Además, «Cell Surface Receiver Signaling Pathway Involved in Cell-Cell Signaling» era un término funcional significativo, que se refiere a una serie de señales moleculares iniciadas cuando un ligando extracelular se une a un receptor en la superficie celular. A continuación, desencadena una cascada de eventos dentro de la célula y, en última instancia, regula los procesos celulares posteriores, como como el metabolismo de los lípidos. En este contexto, se ha documentado el desarrollo de tejido adiposo a través de la señalización célula-célula (67). Se ha sugerido que BMP2, como gen involucrado en esta vía, está asociado con el desarrollo de preadipocitos específicos de depósito y la expansión de AT abdominal en humanos a través de la vía de señalización BMP2-SMAD1/5/8 (68). El otro gen importante en esta vía es el DDX3X, que se conoce como un potencial regulador clave de la adiposidad (69).
Para confirmar las firmas de selección sugeridas anteriormente e identificar nuevos candidatos, se realizó una revisión exhaustiva de la literatura de estudios similares, centrándose en el análisis de identificación de firmas de selección entre razas de ovejas de cola gorda y fina. Entre los 87 genes en las regiones seleccionadas (con símbolos genéticos), 33 genes fueron reportados como genes candidatos involucrados en el desarrollo de la cola gorda en estudios previos (material suplementario S4). En primer lugar, la consistencia entre nuestros resultados (inferidos a partir de los datos de RNA-Seq) y los hallazgos anteriores (basados en DNA-Seq/Array) enfatizan que los conjuntos de datos de RNA-Seq son beneficiosos para la genética de poblaciones, especialmente para el análisis de identificación de firmas de selección. Por otro lado, entre los genes previamente reportados, BMP2 fue de gran interés, ya que se ha reportado que está asociado con el desarrollo de la cola gorda en la mayoría de los estudios de cohortes previos. Los miembros de la familia de las proteínas morfogenéticas óseas (BMP) son conocidos por su papel crucial en la morfogénesis ósea. Numerosos estudios han destacado el papel de BMP2 en la deposición de grasa, así como en la regulación del metabolismo de la adipogénesis (8, 10, 13, 16, 70-74). En perfecto acuerdo con estos estudios, nuestros resultados enfatizaron el papel potencial de BMP2 como el candidato más prometedor asociado con la morfología de la cola gorda en ovejas. En contraste, el PDGFD se reporta como el factor predominante para el fenotipo de cola gorda en ovejas y no se detecta en nuestros resultados (75). Se puede atribuir a muchas razones, incluida la densidad promedio/baja de los SNP identificados por los datos de RNA-Seq, el tamaño inadecuado de la muestra o los métodos utilizados para detectar las firmas. Investigaciones posteriores de los genes identificados indicaron que la morfología de la cola gorda en las ovejas podría explicarse por diversos mecanismos reguladores. Por lo tanto, vale la pena destacar otros genes importantes que se han reportado como directamente asociados con el metabolismo de los lípidos, como RGMA, IGFBP7, UBR5, WLS y NID2. El RGMA media la expresión del receptor activado por el proliferador de peroxisomas (PPARγ), un modulador clave del metabolismo de los lípidos (76). Además, RGMA está asociado con la vía de señalización BMP. Se ha demostrado que esta vía es relevante tanto para la adipogénesis blanca como para la marrón (77). Se ha descrito el papel de IGFBP7 como regulador del metabolismo anormal de los lípidos (78). UBR5 y WLS están implicados en la señalización de Wnt, que es un conocido y principal regulador de la adipogénesis (79). Se ha sugerido que NID2 puede regular la adipogénesis y la distribución de la grasa corporal (80). Estos hallazgos mostraron claramente que el efecto de la selección sobre los genes implicados en la beta-oxidación de los ácidos grasos no es la única forma en que la selección moldeó el perfil genético de las ovejas para adaptarse a diversas y a menudo condiciones de escasez de alimentos. Por lo tanto, los genes descritos anteriormente, especialmente BMP2, pueden considerarse candidatos involucrados en el desarrollo de la cola gorda durante la evolución de las razas ovinas y también pueden considerarse como base para estudios posteriores de rasgos relacionados.
El análisis de PCA mostró una separación robusta entre los dos grupos de ovejas, revelando un alto grado de similitud entre las muestras dentro de cada grupo (grupos de cola gorda y delgada). Esta clara separación entre los dos grupos puede sugerir un origen genético común para cada grupo. En este contexto, está bien establecido que el ancestro salvaje de las ovejas domésticas, la oveja muflón, es de cola fina. En consecuencia, se espera que las ovejas modernas de cola delgada muestren el estado alélico ancestral, mientras que las ovejas de cola gorda exhiben alelos derivados en loci asociados con el fenotipo de cola gorda (75). Sobre la base de la varianza explicada por cada PC, se encontró que 629 y seis SNP importantes contribuían a PC1 y PC2 y estaban relacionados con 70 y dos genes, respectivamente. Se anotó directamente que varios de estos genes (incluidos CYP4V2, IKBKG, BDH2, BMP2, ERBIN y ECHS1) estaban involucrados en los procesos biológicos relacionados con el metabolismo de las grasas, y se informó que 29 genes estaban involucrados en la deposición de grasa según estudios previos. Por ejemplo, el CYP4V2 codifica una enzima que produce ácido araquidónico y desempeña un papel importante en la deposición de grasa. Este gen influye en el almacenamiento de grasa abdominal en bovinos y cerdos (81, 82). Además, un estudio reciente describió el papel de CYP4V2 en la acumulación de gotas de lípidos en las células del epitelio pigmentario de la retina en el pez cebra (83). El gen IKBKG desempeña un papel importante en el metabolismo de las grasas al regular los niveles de lípidos en sangre, la sensibilidad a la insulina y el consumo de energía (84). Estos hallazgos reforzaron los resultados del análisis de identificación de firmas de selección y sugirieron una fuerte selección positiva en estos loci. En conjunto, estos resultados nos permitieron explicar la base genética del tamaño de la cola gorda en las ovejas. Aquí, intentamos discutir los genes candidatos que se informó que estaban asociados con el metabolismo de las grasas. Aunque otros genes candidatos no tienen evidencia directa de participación en el metabolismo de las grasas, son dignos de una investigación más profunda. Finalmente, como esto es una limitación para nuestro estudio, está claro que los datos de RNA-Seq solo capturan SNP dentro de las regiones génicas expresadas, excluyen inherentemente las variantes regulatorias, los intrones y otras regiones no codificantes. Además, un mayor tamaño de la muestra, en particular el mayor número de muestras para cada par de razas, puede mejorar el poder estadístico de los hallazgos, así como la generalización de los resultados, lo que reconocemos como otra limitación de este estudio. Mientras que Rambouillet v2 es la última versión del genoma de referencia de las ovejas en el momento de escribir este artículo, Rambouillet v1 era la última versión en el momento del análisis. Sin embargo, el uso de la nueva versión del genoma no puede cambiar los resultados generales, ya que Rambouillet v2 mejoró la continuidad y no se agregan más secuencias al genoma. De hecho, el número de andamios cambia en la nueva versión y las secuencias genómicas de ambas versiones son las mismas.
5 Conclusión
En este estudio, se llevó a cabo un estudio genómico comparativo (razas de ovejas de cola gorda frente a razas de cola fina) basado en los SNP identificados de los conjuntos de datos de RNA-Seq para descubrir las firmas genómicas que pueden estar relacionadas con el desarrollo de la cola gorda. La consistencia en las firmas de selección de candidatos entre nuestros resultados y los de estudios previos mostró la utilidad de los datos de RNA-Seq para la llamada de SNP, así como para el análisis de firmas de selección. El análisis funcional de los genes en las regiones de selección identificadas puso de relieve nuestra hipótesis previa de que los genes asociados a la oxidación de ácidos grasos pueden modular la deposición de grasa en la cola de las ovejas. Curiosamente y de acuerdo con los informes anteriores, el proceso de angiogénesis también se enriqueció, lo que refuerza su potencial papel regulador en la deposición de grasa de la cola de las ovejas. Además, nuestros hallazgos revelaron que las regiones genómicas identificadas que contienen genes directamente relacionados con el metabolismo de las grasas y también se informó previamente que están asociadas con el fenotipo de cola gorda en ovejas, como BMP2, NID2, IKBKG, RGMA, IGFBP7, UBR5, VEGFD y WLS. Además, se identificaron algunos genes conocidos asociados al metabolismo de las grasas, diferentes a los reportados en estudios previos, incluyendo BDH2, ECHS1, AUH, ERBIN y CYP4V2. Los resultados del análisis de enriquecimiento funcional, junto con el PCA, proporcionaron evidencia adicional de la importancia de las regiones identificadas en el desarrollo de la cola grasa. Estos hallazgos sugieren que las firmas de selección identificadas pueden considerarse como fuerzas evolutivas específicas responsables de la adaptación de la raza de cola gorda a diversas condiciones ambientales, lo que también ha llevado a una mejor comprensión de la evolución de las ovejas en diferentes especies.
Declaración de disponibilidad de datos
Los conjuntos de datos presentados en este estudio se pueden encontrar en repositorios en línea. Los nombres de los repositorios y los números de acceso se pueden encontrar en el artículo/Material complementario.
Declaración ética
El estudio en animales fue aprobado por el Público en el que se utilizaron conjuntos de datos. El estudio se llevó a cabo de acuerdo con la legislación local y los requisitos institucionales.
Contribuciones de los autores
HA: Investigación, Metodología, Visualización, Redacción – borrador original. MB: Conceptualización, Análisis formal, Investigación, Metodología, Visualización, Escritura – borrador original, Escritura – revisión y edición. MM: Metodología, Redacción – revisión y edición. JM: Metodología, Redacción – revisión y edición.
Financiación
El/los autor/es declara(n) que no se recibió apoyo financiero para la investigación, autoría y/o publicación de este artículo.
Conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un posible conflicto de intereses.
El autor o autores declararon ser miembros del comité editorial de Frontiers, en el momento de la presentación. Esto no tuvo ningún impacto en el proceso de revisión por pares ni en la decisión final.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o afirmación que pueda hacer su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fvets.2024.1415027/full#supplementary-material
Referencias
1. Taylor, WTT, Pruvost, M, Posth, C, Rendu, W, Krajcarz, MT, Abdykanova, A, et al. Evidencia de la dispersión temprana de ovejas domésticas en Asia Central. Nat Hum se comporta. (2021) 5:1169–79. DOI: 10.1038/s41562-021-01083-Y
Resumen de PubMed | Texto completo de Crossref | Google Académico
2. Meadows, JRS, Chan, EKF y Kijas, JW. Desequilibrio de ligamiento comparado entre cinco poblaciones de ovejas domésticas. BMC Genet. (2008) 9:1–10. doi: 10-1186/1471-2156-9-61/TABLES/4
3. Kalds, P, Huang, S, Chen, Y y Wang, X. OVINE HOXB13: expansión del repertorio genético del patrón de cola de oveja e implicaciones en la mejora genética. Commun Biol. (2022) 5:1196. doi: 10.1038/s42003-022-04199-7
Resumen de PubMed | Texto completo de Crossref | Google Académico
4. Bakhtiarizadeh, MR, y Alamouti, AA. El descubrimiento de variantes genéticas basadas en RNA-Seq proporciona nuevos conocimientos sobre el control de la deposición de grasa en la cola de las ovejas. Informes de ciencia. (2020) 10:13525. doi: 10.1038/s41598-020-70527-8
Resumen de PubMed | Texto completo de Crossref | Google Académico
5. Oleksyk, TK, Smith, MW, y O’Brien, SJ. Escaneos de todo el genoma en busca de huellas de selección natural. Philos Trans R Soc B Biol Sci. (2010) 365:185–205. doi: 10.1098/RSTB.2009.0219
Resumen de PubMed | Texto completo de Crossref | Google Académico
6. Esteban, W . Barridos selectivos. Genética. (2019) 211:5–13. doi: 10.1534/GENÉTICA.118.301319
Resumen de PubMed | Texto completo de Crossref | Google Académico
7. Horscroft, C, Ennis, S, Pengelly, RJ, Sluckin, TJ y Collins, A. Métodos de la era de la secuenciación para identificar firmas de selección en el genoma. Breve Bioinforme. (2019) 20:1997–2008. doi: 10.1093/BIB/BBY064
Resumen de PubMed | Texto completo de Crossref | Google Académico
8. Zhao, F, Deng, T, Shi, L, Wang, W, Zhang, Q, Du, L, et al. El escaneo genómico para la firma de selección revela la deposición de grasa en ovejas indígenas chinas con tipos de cola extremos. Animales. (2020) 10:773. doi: 10.3390/ani10050000
9. Yuan, Z, Liu, E, Liu, Z, Kijas, JW, Zhu, C, Hu, S, et al. El análisis de la firma de selección revela genes asociados con el tipo de cola en las ovejas indígenas chinas. Anim Genet. (2017) 48:55–66. doi: 10.1111/AGE.12477
10. Moradi, MH, Nejati-Javaremi, A, Moradi-Shahrbabak, M, Dodds, KG, Brauning, R, y McEwan, JC. El mapeo a dedo de las regiones candidatas asociadas con la deposición de grasa en las razas de ovejas iraníes de cola delgada y gorda sugiere nuevos conocimientos sobre los aspectos moleculares de la selección de la cola gorda. Animales. (2022) 12:1423. doi: 10.3390/ani12111423
Resumen de PubMed | Texto completo de Crossref | Google Académico
11. Moradi, MH, Nejati-Javaremi, A, Moradi-Shahrbabak, M, Dodds, KG, y McEwan, JC. Escaneo genómico de barridos selectivos en razas de ovejas de cola delgada y gorda para la identificación de regiones candidatas asociadas con la deposición de grasa. BMC Genet. (2012) 13:10. doi: 10.1186/1471-2156-13-10
Resumen de PubMed | Texto completo de Crossref | Google Académico
12. Manzari, Z, Mehrabani-Yeganeh, H, Nejati-Javaremi, A, Moradi, MH y Gholizadeh, M. Detección de firmas de selección en tres razas de ovejas iraníes. Anim Genet. (2019) 50:298–302. doi: 10.1111/AGE.12772
Resumen de PubMed | Texto completo de Crossref | Google Académico
13. Mastrangelo, S, Moioli, B, Ahbara, A, Latairish, S, Portolano, B, Pilla, F, et al. El escaneo de todo el genoma de las ovejas de cola gorda identifica señales de selección para la deposición y adaptación de grasa. Anim Prod Sci. (2018) 59:835–48. DOI: 10.1071/AN17753
14. Al-Mamun, HA, Kwan, P, Clark, SA, Ferdosi, MH, Tellam, R y Gondro, C. El estudio de asociación de todo el genoma del peso corporal en ovejas merinas australianas revela una región ortóloga en OAR6 a regiones genómicas humanas y bovinas que afectan la altura y el peso. Genet Sel Evol. (2015) 47:66. doi: 10.1186/s12711-015-0142-4
Resumen de PubMed | Texto completo de Crossref | Google Académico
15. Shao, J, Pan, X, Yang, Z, Nanaei, HA, Chen, L, Li, R, et al. La expresión aleloespecífica revela las diferencias fenotípicas entre las ovejas de cola delgada y las de cola gorda. J Genómica de Genet. (2020) 49:583–6. doi: 10.21203/RS.3.RS-56388/V1
16. Lv, X, Chen, W, Wang, S, Cao, X, Yuan, Z, Getachew, T, et al. Resecuenciación del genoma completo de las ovejas Dorper y Hu para revelar firmas de selección asociadas con rasgos importantes. Anim Biotechnol. (2023) 34:3016–26. doi: 10.1080/10495398.2022.2127409
17. Ahbara, A, Bahbahani, H, Almathen, F, Al, AM, Agoub, MO, Abeba, A, et al. Variación de todo el genoma, regiones candidatas y genes asociados con la deposición de grasa y la morfología de la cola en ovejas indígenas etíopes. Frente Genet. (2019) 10:387968. doi: 10.3389/FGENE.2018.00699/BIBTEX
18. Satam, H, Joshi, K, Mangrolia, U, Waghoo, S, Zaidi, G, Rawool, S, et al. Tecnología de secuenciación de última generación: tendencias y avances actuales. Biol. (2023) 12:997. doi: 10.3390/BIOLOGY12070997
19. Xu, SS, Gao, L, Shen, M y Lyu, F. Los escaneos selectivos del genoma completo detectan genes asociados con rasgos fenotípicos importantes en ovejas (Ovis aries). Frente Genet. (2021) 12:738879. doi: 10.3389/fgene.2021.738879
Resumen de PubMed | Texto completo de Crossref | Google Académico
20. Piskol, R, Ramaswami, G, y Li, JB. Identificación fiable de variantes genómicas a partir de datos de RNA-Seq. Am J Hum Genet. (2013) 93:641–51. doi: 10.1016/J.AJHG.2013.08.008
Resumen de PubMed | Texto completo de Crossref | Google Académico
21. Bakhtiarizadeh, MR, Salehi, A, Alamouti, AA, Abdollahi-Arpanahi, R y Salami, A. El análisis profundo del transcriptoma mediante RNA-seq sugiere nuevos conocimientos sobre los aspectos moleculares del metabolismo de la cola gorda en las ovejas. Sci Rep. 9:9203. doi: 10.1038/s41598-019-45665-3
22. Guangli, Y, Huan, Z, Shuhong, Z, Zhiqiang, L, Fengyi, G, Guan, W, et al., Identificación de la base genética para el rasgo fenotípico de cola grande en ovejas Han a través del análisis integrado de ARNm y miARN de muestras de tejido graso de la cola. (2020). Disponible en: https://www.researchsquare.com/article/rs-107294/v1
23. Ma, L, Zhang, M, Jin, Y, Erdenee, S, Hu, L, Chen, H, et al. Perfil comparativo del transcriptoma de ARNm y ARNc relacionados con los tejidos adiposos de la cola de ovejas. Frente Genet. (2018) 9:355770. doi: 10.3389/FGENE.2018.00365/BIBTEX
24. Yuan, Z, Ge, L, Sun, J, Zhang, W, Wang, S, Cao, X, et al. El análisis integrador de los datos de Iso-Seq y RNA-seq revela la complejidad del transcriptoma y las transcripciones expresadas diferencialmente en la grasa de la cola de oveja. ParesJ. (2021) 9:E12454. doi: 10.7717/peerj.12454
Resumen de PubMed | Texto completo de Crossref | Google Académico
25. Fan, H, Hou, Y, Sahana, G, Gao, H, Zhu, C, Du, L, et al. Estudio transcriptómico de la deposición de grasa de la cola en dos tipos de ovejas Hulun Buir según el tamaño de la cola y el sexo. Animal. (2019) 9:655. doi: 10.3390/ANI9090655
26. Farhadi, S, Ghias, JS, Hasanpur, K, Mohammadi, SA y Ebrahimie, E. Mecanismos moleculares de la deposición de grasa: IL-6 es un gen central en la lipólisis de grasa, que compara razas de ovejas de cola fina con de cola gorda. Raza Arch Anim. (2021) 64:53–68. doi: 10.5194/AAB-64-53-2021
Resumen de PubMed | Texto completo de Crossref | Google Académico
27. Jin, M, Fei, X, Li, T, Lu, Z, Chu, M, Di, R, et al. Oar-miR-432 regula la diferenciación de grasas y promueve la expresión de BMP2 en preadipocitos ovinos. Frente Genet. (2022) 13:844747. doi: 10.3389/fgene.2022.844747
Resumen de PubMed | Texto completo de Crossref | Google Académico
28. Brown, J, Pirrung, M y Mccue, LA. Panel de control FQC: integra los resultados de FastQC en una herramienta de control de calidad FASTQ basada en la web, interactiva y extensible. Bioinformática. (2017) 33:3137–9. doi: 10.1093/BIOINFORMATICS/BTX373
Resumen de PubMed | Texto completo de Crossref | Google Académico
29. Bolger, AM, Lohse, M y Usadel, B. Trimmomatic: un recortador flexible para los datos de secuencia de Illumina. Bioinformática. (2014) 30:2114–20. doi: 10.1093/BIOINFORMÁTICA/BTU170
Resumen de PubMed | Texto completo de Crossref | Google Académico
30. Veeneman, BA, Shukla, S, Dhanasekaran, SM, Chinnaiyan, AM, y Nesvizhskii, AI. La alineación de dos pasos mejora la cuantificación de las uniones de empalme novedosas. Bioinformática. (2016) 32:43–9. doi: 10.1093/BIOINFORMATICS/BTV642
Resumen de PubMed | Texto completo de Crossref | Google Académico
31. Hosseini, SF, Bakhtiarizadeh, MR y Salehi, A. El metanálisis de los conjuntos de datos de RNA-Seq destaca nuevos genes/vías implicados en la deposición de grasa en la cola de grasa de las ovejas. Frente Vet Sci. (2023) 10:1159921. doi: 10.3389/fvets.2023.1159921
Resumen de PubMed | Texto completo de Crossref | Google Académico
32. Bathke, J y Lühken, G. OVarFlow: una variante de código abierto basada en GATK 4 optimizada para recursos que llama a workFlow. BMC Bioinform. (2021) 22:1–18. doi: 10.1186/S12859-021-04317-Y/FIGURES/4
33. Ebbert, MTW, Wadsworth, ME, Staley, LA, Hoyt, KL, Pickett, B, Miller, J, et al. Evaluar la necesidad de eliminar los duplicados de PCR de los datos de secuenciación de próxima generación y una comparación de enfoques. BMC Bioinform. (2016) 17:491–500. doi: 10.1186/S12859-016-1097-3/FIGURES/6
34. Deng, J, Xie, XL, Wang, DF, Zhao, C, Lv, FH, Li, X, et al. Orígenes paternos y episodios migratorios de las ovejas domésticas. Curr Biol. (2020) 30:4085–4095.e6. doi: 10.1016/J.CUB.2020.07.077
Resumen de PubMed | Texto completo de Crossref | Google Académico
35. Yang, W, Yang, Y, Zhao, C, Yang, K, Wang, D, Yang, J, et al. Animal-ImputeDB: una base de datos completa con múltiples paneles de referencia de animales para la imputación de genotipos. Ácidos nucleicos Res. (2020) 48:D659–67. doi: 10.1093/NAR/GKZ854
Resumen de PubMed | Texto completo de Crossref | Google Académico
36. Elevación . Wiki de Análisis del Genoma (2023). Disponible en: https://genome.sph.umich.edu/wiki/LiftOver (consultado el 19 de diciembre de 2023)
37. Browning, BL, Tian, X, Zhou, Y y Browning, SR. Fase rápida en dos etapas de datos de secuencia a gran escala. Am J Hum Genet. (2021) 108:1880–90. doi: 10.1016/J.AJHG.2021.08.005
38. Browning, BL, Zhou, Y y Browning, SR. Un genoma imputado de un centavo de paneles de referencia de próxima generación. Am J Hum Genet. (2018) 103:338–48. doi: 10.1016/J.AJHG.2018.07.015
Resumen de PubMed | Texto completo de Crossref | Google Académico
39. Yun, L, Willer, C, Sanna, S y Abecasis, G. Imputación de genotipos. Annu Rev Genomics Hum Genet. (2009) 10:387–406. doi: 10.1146/ANNUREV. GENOM.9.081307.164242
40. Marchini, J y Howie, B. Imputación de genotipos para estudios de asociación de todo el genoma. Nat Rev Genet. (2010) 11:499–511. DOI: 10.1038/NRG2796
41. Yin, Y, Hou, L, Liu, C, Li, K, Guo, H, Niu, P, et al. El estudio de asociación de todo el genoma identificó un locus de rasgo cuantitativo y dos genes candidatos en el cromosoma 2 de Sus scrofa que afectan los rasgos vulvares de los cerdos Suhuai. Genes (Basilea). (2022) 13:1294. doi: 10.3390/genes13081294
Resumen de PubMed | Texto completo de Crossref | Google Académico
42. Hao, K, Chudin, E, McElwee, J y Schadt, EE. Precisión de la imputación de marcadores no tipificados en todo el genoma e impactos en el poder estadístico para estudios de asociación. BMC Genet. (2009) 10:1–10. doi: 10.1186/1471-2156-10-27/FIGURES/4
43. MacEachern, S, Hayes, B, McEwan, J y Goddard, M. Un examen de la selección positiva y el cambio del tamaño efectivo de la población en las poblaciones de ganado Angus y Holstein (Bos taurus) utilizando una plataforma de genotipado de SNP de alta densidad y la contribución del polimorfismo antiguo a la diversidad genómica en el ganado doméstico. BMC Genómica. (2009) 10:1–19. doi: 10.1186/1471-2164-10-181/TABLES/6
44. Weir, B.B., y Cockerham, C.C. Estimación de estadísticos F para el análisis de la estructura de la población. Evolución (N Y). (1984) 38:1358. doi: 10.2307/2408641
45. Gholami, M, Erbe, M, Gärke, C, Preisinger, R, Weigend, A, Weigend, S, et al. Análisis genómico poblacional basado en 1 millón de SNPs en ponedoras de huevos comerciales. PLoS Uno. (2014) 9:E94509. doi: 10.1371/REVISTA. PONE.0094509
Resumen de PubMed | Texto completo de Crossref | Google Académico
46. Cingolani, P, Platts, A, Wang, LL, Coon, M, Nguyen, T, Wang, L, et al. Un programa para anotar y predecir los efectos de polimorfismos de un solo nucleótido, SnpEff. Mosca (Austin). (2012) 6:80–92. doi: 10.4161/FLY.19695
Resumen de PubMed | Texto completo de Crossref | Google Académico
47. Chen, EY, Tan, CM, Kou, Y, Duan, Q, Wang, Z, Meirelles, GV, et al. Enrichr: herramienta interactiva y colaborativa de análisis de enriquecimiento de listas de genes HTML5. BMC Bioinform. (2013) 14:128. doi: 10.1186/1471-2105-14-128
Resumen de PubMed | Texto completo de Crossref | Google Académico
48. Merkin, J, Russell, C, Chen, P y Burge, CB. Dinámica evolutiva de la regulación génica e isoforme en tejidos de mamíferos. Ciencia. (2012) 338:1593–9. doi: 10.1126/SCIENCE.1228186/SUPPL_FILE/MERKIN. SM.PDF
49. Chan, ET, Quon, GT, Chua, G, Babak, T, Trochesset, M, Zirngibl, RA, et al. Conservación de la expresión génica central en tejidos de vertebrados. J Biol. (2009) 8:1–17. doi: 10.1186/JBIOL130/FIGURES/8
50. Gutiérrez-Gil, B, Arranz, JJ, y Wiener, P. Una revisión interpretativa de los estudios de barrido selectivo en las poblaciones de ganado Bos taurus: identificación de señales de selección únicas y compartidas entre razas. Frente Genet. (2015) 6:167. doi: 10.3389/FGENE.2015.00167/FULL
51. Muigai, AWT, y Hanotte, O. El origen de las ovejas africanas: perspectivas arqueológicas y genéticas. Arqueología Africana Rev. (2013) 30:39–50. doi: 10.1007/s10437-013-9129-0
52. Cadzow, M, Boocock, J, Nguyen, HT, Wilcox, P, Merriman, TR y Black, MA. Un flujo de trabajo bioinformático para detectar firmas de selección en datos genómicos. Frente Genet. (2014) 5:102714. doi: 10.3389/FGENE.2014.00293/RESUMEN
53. Yang, J, Li, WR, Lv, FH, He, SG, Tian, SL, Peng, WF, et al. La secuenciación del genoma completo de ovejas autóctonas proporciona información sobre las adaptaciones rápidas a entornos extremos. Mol Biol Evol. (2016) 33:2576–92. doi: 10.1093/MOLBEV/MSW129
Resumen de PubMed | Texto completo de Crossref | Google Académico
54. Ai, H, Fang, X, Yang, B, Huang, Z, Chen, H, Mao, L, et al. Adaptación y posible introgresión antigua entre especies en cerdos identificados por secuenciación del genoma completo. Nat Genet. (2015) 47:217–25. doi: 10.1038/ng.3199
Resumen de PubMed | Texto completo de Crossref | Google Académico
55. Zhang, S, Yao, Z, Li, X, Zhang, Z, Liu, X, Yang, P, et al. Evaluación de la diversidad genómica y las firmas de selección en el ganado Pinan utilizando datos de secuenciación del genoma completo. BMC Genómica. (2022) 23:1–10. doi: 10-1186/S12864-022-08645-Y/TABLAS/1
56. Shafiei, H, Bakhtiarizadeh, MR y Salehi, A. Perfil de edición de ARN potencial a gran escala en diferentes tejidos de pollo adulto. Anim Genet. (2019) 50:460–74. doi: 10.1111/AGE.12818
Resumen de PubMed | Texto completo de Crossref | Google Académico
57. Bakhtiarizadeh, MR, Salehi, A y Rivera, RM. Identificación y análisis de todo el genoma de eventos de edición de ARN A a I en bovinos mediante secuenciación del transcriptoma. PLoS Uno. (2018) 13:E0193316. doi: 10.1371/REVISTA. PONE.0193316
Resumen de PubMed | Texto completo de Crossref | Google Académico
58. Bonnet, A, Cao, KL, y Sancristóbal, M. Expresión génica in vivo en células de la granulosa durante el desarrollo folicular terminal del cerdo. Reproducción. (2008) 136:211–24. doi: 10.1530/REP-07-0312
59. Kamei, A, Watanabe, Y, Kondo, K, Okada, S, Shinozaki, F, Ishijima, T, et al. Influencia de una dieta deficiente en hierro a corto plazo en los perfiles de expresión génica hepática en ratas. PLoS Uno. (2013) 8:E65732. doi: 10.1371/REVISTA. PONE.0065732
Resumen de PubMed | Texto completo de Crossref | Google Académico
60. Wang, L, Zhang, Y, Zhang, B, Zhong, H, Lu, Y y Zhang, H. Detección de genes candidatos para la deposición de lípidos utilizando datos transcriptómicos y proteómicos combinados de cerdos negros de Nanyang. BMC Genómica. (2021) 22:441. doi: 10.1186/S12864-021-07764-2
Resumen de PubMed | Texto completo de Crossref | Google Académico
61. Peng, M, Han, J, Li, L, e Informes, HM-S. Supresión de la deposición de grasa en pollos de engorde mediante la suplementación con ácido (–hidroxicítrico: una perspectiva proteómica) (2016). Disponible en: https://www.nature.com/articles/srep32580 (consultado el 17 de diciembre de 2023).
62. Fan, C, Hu, H, Huang, X, Su, D, Huang, F, Zhuo, Z, et al. La suplementación con betaína provoca un aumento en la oxidación de ácidos grasos y el metabolismo de los carbohidratos en el hígado de ratones alimentados con una dieta alta en grasas: un análisis proteómico. Seguridad alimentaria. (2022) 11:881. doi: 10.3390/FOODS11060881
63. Alonso-García, M, Suárez-Vega, A, Fonseca, PAS, Marina, H, Pelayo, R, Mateo, J, et al. Análisis del transcriptoma de la grasa perirrenal de las canales de cordero lechal español de Assaf que muestra diferentes niveles de grasa renal y de canal. Frente Vet Sci. (2023) 10:1150996. doi: 10.3389/fvets.2023.1150996
Resumen de PubMed | Texto completo de Crossref | Google Académico
64. Alonso-García, M, Gutiérrez-Gil, B, Pelayo, R, Fonseca, PAS, Marina, H, Arranz, JJ, et al. Un enfoque de metanálisis para la anotación e identificación de lncRNAs que controlan la deposición de grasa perirrenal en corderos lechales. Anim Biotechnol. (2024) 35:2374328. doi: 10.1080/10495398.2024.2374328
Resumen de PubMed | Texto completo de Crossref | Google Académico
65. Lijnen, H, Frederix, L, Van Hoef, B y Dewerchin, M. La deficiencia del factor de crecimiento endotelial vascular-D no afecta el desarrollo del tejido adiposo murino (2009). Disponible en: https://www.sciencedirect.com/science/article/pii/S0006291X08022225 (consultado el 2 de septiembre de 2024).
66. Voros, G, Maquoi, E, Demeulemeester, D, Clerx, N, Collen, D y Lijnen, HR. Modulación de la angiogénesis durante el desarrollo del tejido adiposo en modelos murinos de obesidad. Endocrinología. (2005) 146:4545–54. doi: 10.1210/EN.2005-0532
Resumen de PubMed | Texto completo de Crossref | Google Académico
67. Khan, T, Muise, ES, Iyengar, P, Wang, ZV, Chandalia, M, Abate, N, et al. Desregulación metabólica y fibrosis del tejido adiposo: papel del colágeno VI. Mol Cell Biol. (2009) 29:1575–91. doi: 10.1128/mcb.01300-08
Resumen de PubMed | Texto completo de Crossref | Google Académico
68. Denton, NF, Eghleilib, M, Al-Sharifi, S, Todorčević, M, Neville, MJ, Loh, N, et al. La proteína morfogenética ósea 2 es un regulador específico del depósito de la adipogénesis humana. Int J Obes. (2019) 43:2458–68. doi: 10.1038/s41366-019-0421-1
Resumen de PubMed | Texto completo de Crossref | Google Académico
69. Savva, C, Helguero, LA, González-Granillo, M, Melo, T, Couto, D, Buyandelger, B, et al. La dieta materna alta en grasas programa el lipidoma y el transcriptoma del tejido adiposo blanco y marrón en la descendencia de una manera dependiente del sexo y el tejido en ratones. Int J Obes. (2022) 46:831–42. doi: 10.1038/s41366-021-01060-5
Resumen de PubMed | Texto completo de Crossref | Google Académico
70. Moioli, B, Pilla, F y Ciani, E. Las firmas de selección identifican loci asociados con la cola gorda en las ovejas. J Anim Sci. (2015) 93:4660–9. doi: 10.2527/jas.2015-9389
71. Deniskova, T, Kunz, E, y Medugorac, I. Un estudio de los mecanismos genéticos que subyacen al fenotipo de la cola gorda en ovejas: enfoques metodológicos y genes candidatos identificados (2019) Disponible en: http://www.agrobiology.ru/articles/6-2019deniskova.pdf (consultado el 17 de diciembre de 2023).
72. Zhang, C, Zhang, J, Tuersuntuoheti, M, Zhou, W, Han, Z, Li, X, et al. La genómica del paisaje revela la divergencia adaptativa de las ovejas autóctonas en diferentes entornos ecológicos de Xinjiang, China. Sci Total Environ. (2023) 904:166698. doi: 10.1016/j.scitotenv.2023.166698
73. Bedhiaf-Romdhani, S, Baazaoui, I, Dodds, KG, Brauning, R, Anderson, RM, Van Stijn, TC, et al. Eficiencia del genotipado por secuenciación para inferir la relación genómica y los conocimientos moleculares sobre la selección de la cola gorda en ovejas tunecinas. Anim Genet. (2023) 54:389–97. doi: 10.1111/AGE.13296
Resumen de PubMed | Texto completo de Crossref | Google Académico
74. Baazaoui, I, Bedhiaf-Romdhani, S, y Animal, SM. Los análisis de todo el genoma revelan la estructura de la población e identifican genes candidatos asociados con la grasa de la cola en ovejas locales de una zona semiárida. Animal. (2021) 15:100193. doi: 10.1016/j.animal.2021.100193
75. Dong, K, Yang, M, Han, J, Ma, Q, Han, J, Song, Z, et al. El análisis genómico de las razas ovinas de todo el mundo revela que el PDGFD es uno de los principales objetivos de la selección de cola gorda en las ovejas. BMC Genómica. (2020) 21:1–12. doi: 10.1186/S12864-020-07210-9/FIGURES/4
76. Yuan, H, Suzuki, S, Hirata-Tsuchiya, S, Sato, A, Nemoto, E, Saito, M, et al. La acetilación global de H3K27 inducida por PPARγ mantiene las capacidades osteo/cementogénicas de los fibroblastos del ligamento periodontal. Int J Mol Sci. (2021) 22:8646. doi: 10.3390/ijms22168646
77. Mir, FA, Mall, R, Iskandarani, A, Ullah, E, Samra, TA, Cyprian, F, et al. MicroARN característicos vinculados a vías metabólicas desreguladas en sujetos adultos qataríes con obesidad y síndrome metabólico. Endocrinol frontal (Lausana). (2022) 13:937089. doi: 10.3389/fendo.2022.937089
Resumen de PubMed | Texto completo de Crossref | Google Académico
78. Stanley, T, Fourman, L, McClure, CM, Feldpausch, MN, Torriani, M, y Corey, KE. Relación del IGF-1 y las proteínas de unión al IGF con la gravedad de la enfermedad y la glucemia en la enfermedad del hígado graso no alcohólico. J Clin Endocrinol Metab. (2021) 106:E520–33. doi: 10.1210/clinem/dgaa792
79. Song, K, Wang, S, Mani, M y Mani, A. Señalización de Wnt, lipogénesis de novo, adipogénesis y grasa ectópica. Oncotarget. (2014) 5:11000–3. doi: 10.18632/oncotarget.2769
80. Sol, C, Kovacs, P y genes, EG-J. Genética de la distribución de la grasa corporal: análisis comparativos en poblaciones con ascendencia europea, asiática y africana. Genes (Basilea). (2021, 2021) 12:841. doi: 10.3390/genes12060841
81. Zhang, Y, Sun, Y, Wu, Z, Xiong, X, Zhang, J, Ma, J, et al. Los transcriptomas de grasa subcutánea e intramuscular muestran grandes diferencias en la organización de la red y asociaciones con los rasgos adiposos en los cerdos. Sci China Life Sci. (2021) 64:1732–46. doi: 10.1007/s11427-020-1824-7
Resumen de PubMed | Texto completo de Crossref | Google Académico
82. Schumacher, ML, Boles, JA, y Thomson, JM. Un enfoque comparativo para refinar los mecanismos moleculares que influyen en la calidad de la carne y las características de la canal. Transl Anim Sci. (2021) 5:S189-94. doi: 10.1093/TAS/TXAB184
83. Gao, P, Jia, D, Li, P, Huang, Y, Hu, H, Sun, K, et al. Acumulación de gotas de lípidos en un nuevo modelo de pez cebra con distrofia cristalina de Bietti con alteración de la vía PPARα. Invertir Ophthalmol Vis Sci. (2022) 63:32–2. doi: 10.1167/IOVS.63.5.32
Resumen de PubMed | Texto completo de Crossref | Google Académico
Palabras clave: Conjuntos de datos de RNA-Seq, firmas de selección, deposición de grasa, ovejas de cola fina y gorda, llamadas de SNP
Cita: Abbasabadi H, Bakhtiarizadeh MR, Moradi MH y McEwan JC (2024) Análisis de firma de selección basado en RNA-Seq para identificar huellas genómicas asociadas con el fenotipo de cola gorda en ovejas. Frente. Vet. Sci. 11:1415027. doi: 10.3389/fvets.2024.1415027
Editado por:
Beatriz Gutiérrez-Gil, Universidad de León, España
Revisado por:
Tara G. McDaneld, Servicio de Investigación Agrícola (USDA), Estados
Unidos Pamela Wiener, Universidad de Edimburgo, Reino Unido
Mohammad Hossein Banabazi, Universidad Sueca de Ciencias Agrícolas, Suecia
Derechos de autor © 2024 Abbasabadi, Bakhtiarizadeh, Moradi y McEwan. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia Creative Commons Atribución (CC BY).
*Correspondencia: Mohammad Reza Bakhtiarizadeh, mrbakhtiari@ut.ac.ir
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente a las de sus organizaciones afiliadas, o las del editor, de los editores y de los revisores. Cualquier producto que puede ser evaluada en este artículo o afirmación que puede ser hecha por su El fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía