
Características genéticas del subgenotipo 1c del virus de la diarrea viral bovina (BVDV) en terneros recién nacidos en usos de nucleótidos y codones sinónimos

Características genéticas del subgenotipo 1c del virus de la diarrea viral bovina (BVDV) en terneros recién nacidos en usos de nucleótidos y codones sinónimos
- 1Laboratorio Clave de Biotecnología y Bioingeniería de la Comisión Estatal de Asuntos Étnicos, Centro de Investigación Biomédica, Universidad del Noroeste de Minzu, Lanzhou, China
- 2Gansu Tech Innovation Center of Animal Cell, Centro de Investigación Biomédica, Universidad Northwest Minzu, Lanzhou, China
- 3Colegio de Ciencias de la Vida e Ingeniería, Universidad del Noroeste de Minzu, Lanzhou, China
- 4Qinghai Centro Provincial de Control y Prevención de Enfermedades Animales, Xining, China
El virus de la diarrea viral bovina (BVDV), que sirve como un patógeno importante para los terneros recién nacidos, representa una amenaza para las pérdidas reproductivas y económicas en la industria ganadera. Para estudiar la tasa de infección y la diversidad genética de BVDV en terneros recién nacidos en el norte de China, se recogieron un total de 676 muestras de sueros de terneros recién nacidos de cuatro provincias entre 2021 y 2022. Todas las muestras de suero se detectaron individualmente para la infección por BVDV mediante RT-PCR y ELISA. Nuestros resultados mostraron que la tasa serológica general fue del 9,76 % (66/676) y la tasa media positiva de ARN BVDV fue del 8,14 % (55/676) en los terneros recién nacidos. Ocho cepas de BVDV se aislaron con éxito de muestras de suero RT-PCR positivos, y cuatro aislados mostraron el efecto citopático (CPE). Basándose en el árbol filogenético a nivel del genoma, las ocho cepas se clasificaron en el subgenotipo 1c. Además, los aislados de BVDV tenían una estrecha relación genética con la cepa GSTZ a nivel de uso de nucleótidos o codones. Curiosamente, en comparación con los patrones de uso de codones sinónimos entre los aislados de BVDV con CPE y los que no tienen CPE, hubo cuatro codones sinónimos (UCG, CCC, GCA y AAC) que mostraban las diferencias significativas (p < 0,05) en el patrón de uso de codones, lo que sugiere que el sesgo de uso de codones sinónimos podría desempeñar Además, el uso de codones sinónimos que contienen dinucleótidos de CpG fue suprimido por los aislados de BVDV-1c, lo que refleja una de las estrategias de evasión inmune de BVDV a su huésped. En conjunto, nuestro estudio proporcionó datos para el seguimiento y las estrategias de vacunación de BVDV para terneros recién nacidos en el norte de China.
Introducción
Virus de la diarrea viral bovina (BVDV), que sirve como patógeno infeccioso asociado con enfermedades reproductivas, gastrointestinales e infertilidad de la industria ganadera en el mundo. BVDV es un virus de ARN sensorial positivo de una sola cadena envuelto, y se clasifica en el género Pestivirus, familia Flaviviridae. En el genoma BVDV, la región no traducida de 5 ′ (UTR) y la UTR de 3 pulgadas flanquean un marco de lectura abierto (ORF). Dentro del género Pestivirus de la familia Flaviviridae, tres genotipos virales están asociados con la diarrea viral bovina: BVDV-1, BVDV-2 y Pestivirus H (pantevirus similar a HoBi, BVDV-3) (1). La BVDV tiene la capacidad de cruzar la placenta durante el embarazo temprano y, además, conduce al nacimiento de terneros persistentemente infectados (PI) (1). La transmisión vertical de BVDV ocurre cuando el virus se transmite desde la presa infectada a su descendencia. Durante la fase de gestación, la infección fetal puede causar abortos, mortinatos, efectos teratogénicos, además, cuando la infección ocurre durante el primer trimestre, a menudo resulta en el nacimiento de terneros inmunotolerantes, infectados persistentemente, virémicos (2, 3). La inmunotolerancia humoral y celular a las cepas BVDV es una caracterización única de la infección persistente. Debido a la naturaleza de la infección persistente de BVDV, no hay un tratamiento eficaz para curar completamente a un animal con infección por BVDV (4). Por lo tanto, los programas eficaces de control de BVDV representan que la eliminación de animales IP (como los terneros recién nacidos de PI) conduce a la extinción viral en los rebaños de ganado. En general, las secuencias del genoma de los pestivirus rumiantes cambian poco durante el IP. Sin embargo, exhiben una gran heterogeneidad, lo que refleja una larga historia de coevolución virus-huésped en la que las cepas avirulentas tienen más éxito (5).
Los análisis epidemiológicos indican claramente que los factores demográficos, como el tamaño y la densidad del rebaño, se consideran importantes predictores de la prevalencia de la infección en poblaciones donde la BVDV es endémica (6-8). Debido a la naturaleza propensa a errores de las ARN polimerasas responsables de la replicación del genoma viral, BVDV muestra una alta mutación para adaptarse a su huésped susceptible. BVDV-1 es el genotipo dominante en todo el mundo, y se han propuesto al menos 21 subgenotipos (BVDV-1a a BVDV-1u) (9, 10). La infección por BVDV se ha extendido ampliamente por toda China con alta prevalencia y diversidad genética. Con el desarrollo de la industria ganadera en China, siempre se observan diferentes variantes virales y las altas seroprevalencias correspondientes para la infección por BVDV en rebaños de ganado en diferentes provincias (11-16). En particular, la seroprevalencia de BVDV en el ganado lechero es alta, acercándose al 57 %, con una tasa positiva de ARN BVDV del 27,1 % (13). En cuanto a las variantes de BVDV circulan en China, hay al menos 11 subgenotipos de variantes de BVDV que infectan a diferentes animales domésticos, incluyendo BVDV-1a a BVDV-1d, BVDV-1m a BVDV-1q, BVDV-1u, BVDV-2a y BVDV-2b) (17-23). Sin embargo, los estudios anteriores se llevaron a cabo principalmente en rebaños de ganado adulto en China y la prevalencia de la infección por BVDV en terneros recién nacidos parece ser ignorada para las investigaciones. Dado que los terneros recién nacidos con infección por BVDV tienen un deterioro grave para el desarrollo de la industria ganadera en China, los estudios moleculares y serológicos para terneros recién nacidos con infección por BVDV pueden mostrar información valiosa sobre las cepas de BVDV que circulan en una población y, por lo tanto, llevar a cabo un programa de control efectivo, guiar la estrategia de En este estudio, realizamos una encuesta sobre la evaluación de la prevalencia de la infección por BVDV y la diversidad genética de los aislados de BVDV en terneros recién nacidos en el norte de China.
Materiales y métodos
Recogida de muestras de sangre para terneros recién nacidos
Este estudio fue aprobado por el Comité de Ética Animal de la Facultad de Ciencias de la Vida e Ingeniería de la Universidad Northwest Minzu (Aprobación No. CLSEAEC 2020-003). Los terneros recién nacidos dentro de los 14 días de nacimiento se manejaron de acuerdo con las buenas prácticas animales requeridas por los Procedimientos y Directrices de Ética Animal de la República Popular China. De las cuatro provincias (Ningxia, Shanxi, Shandong y Mongolia Interior) de China, los terneros recién nacidos fueron seleccionados al azar, después de obtener el consentimiento verbal de los propietarios de la granja de ganado. Finalmente, se recogieron un total de 676 muestras de sangre, y la operación convencional llevó a cabo la extracción de suero (24). Las muestras de suero se mantuvieron a -20 °C hasta un análisis adicional.
Detección serológica de la infección por BVDV
Todas las muestras de suero para anticuerpos totales contra BVDV se probaron utilizando un kit de prueba ELISA de competición (Ingenasa, España). En cuanto al kit ELISA de la competencia, el péptido recombinante del antígeno p80/p125 del pestivirus se utiliza para el antígeno viral.
Detección de RT-PCR y aislamiento de virus
El ARN total se extrajo de cada muestra de suero utilizando el kit de ADN/ARN de TIANampvirus (TIANGEN Biotech, China). El ARN extraído se transcribió inversamente utilizando el kit de transcriptasa inversa M-MLV (TaKaRa, China) siguiendo al fabricante. Se diseñaron doce juegos de imprimaciones para amplificar las regiones superpuestas que cubren todo el genoma de BVDV (Tabla complementaria 1). El juego de imprimación para amplificar la secuencia 5’UTR se utilizó para detectar preliminarmente el ARN BVDV. Después de la purificación del gel, el producto de amplificación fue secuenciado directamente por Tsingke Biotechnology Co., Ltd.
La muestra de suero positiva, que contenía ARN BVDV, se inoculó en cultivos de monocapa MDBK, frascos de 25 cm con un inóculo de 1 ml (dilución de 1:10 de las muestras originales) y el volumen total fue de 5 ml. Los cultivos celulares se congelaron y descongelaron tres veces y pasaron diez veces a intervalos de 5 días. Era necesario observar el cultivo MDBK procesado para identificar la ausencia o presencia del efecto citopático (ECP). Además, para estimar el título viral de la cepa BVDV con CPE, se utilizó el método de la dosis de infectividad del 50% del cultivo de tejidos (TCID50) para la evaluación correspondiente del título de BVDV (25, 26).
Análisis de las características genéticas de los aislados de BVDV
Análisis filogenético
Para aclarar la relación genética entre los aislados de BVDV presentados y las cepas de BVDV que circulan en China, en este estudio se utilizaron 28 cepas con todo el genoma viral registrado en GenBank (Tabla complementaria 2). Se compiló la reconstrucción filogenética para el genotipado genético de los aislados de BVDV. El árbol filogenético se infiró por el genoma viral produciendo árboles de máxima probabilidad (ML) con el modelo Tamura-Nei más la distribución gamma de la sustitución de la base utilizando el software MAGE 7.0. Aquí, seleccionamos el modelo GTR+G+I para realizar el árbol ML para mostrar la diversidad genética de las cepas BVDV objetivo, de acuerdo con el mínimo del criterio de información bayesiana (BIC), que corresponde a los ajustes de máxima probabilidad del modelo de sustitución de nucleótidos específico.
Análisis de patrones sinónimos de uso de codones para los aislados de BVDV
El valor de uso de codones sinónimos relativo (RSCU) para un codón sinónimo es la relación entre su frecuencia observada y su frecuencia esperada sin uso sesgado (la frecuencia media de todos los miembros sinónimos que codifican su aminoácido específico) (27). RSCU sería una representación objetiva del sesgo de uso de codones sinónimos sin confundir la influencia de la composición de aminoácidos para diferentes cepas de BVDV. En general, cuando el valor de RSCU es superior a 1,0, el codón sinónimo correspondiente muestra una tendencia positiva de uso de codones. Por el contrario, cuando el valor de RSCU es <1,0, el codón sinónimo correspondiente refleja una tendencia negativa de uso de codones (28, 29). Para identificar el alcance de 59 sesgos sinónimos de uso de codones, los valores de RSCU superiores a 1,6 y <0,6 se definieron como codones «sobrerrepresentados» y «subrepresentados», respectivamente (30).
Además, el análisis de componentes principales (PCA) es un método estadístico multivariable que reduce la dimensionalidad de los datos mediante la realización de un análisis de covarianza para una matriz de datos (31). En cuanto a la diversidad genética de los aislados de BVDV con respecto al uso general de codones, se realizó el análisis de PCA para los datos de RSCU de ORF viral. El gráfico que mostraba la diversidad genética de estas cepas se compuso de la primera variante (datos f1‘) y la segunda variante (datos de f2‘), revelando el patrón general de uso de codones derivado de las 59 variantes de uso de codones sinónimos para cada cepa en este estudio.
Análisis estadístico
Se utilizó el método ANOVA unidireccional para comparar las medias de dos o más grupos que contenían datos de respuesta numérica utilizando el software SPSS 16.0 para Windows, y se puede identificar una diferencia significativa cuando el valor de p era < 0,05. La regresión lineal se utilizó para modelar la relación entre una variable dependiente escalar y una variable independiente utilizando el software GraphPad Prism 6 para Windows.
Resultados
Resultados positivos representados por ELISA y RT-PCR para terneros recién nacidos con infección por BVDV
Para estimar la prevalencia serológica de terneros recién nacidos con infección por BVDV, se probó cada muestra de suero para detectar los anticuerpos anti-BVDV. La tasa serológica general fue del 9,76% (66/676) para los terneros recién nacidos en este estudio. Va del 4 al 16 % en el norte de China (Tabla 1). Entre las cuatro provincias, la tasa serológica de infección por BVDV fue más alta en la provincia de Shandong, seguida de Mongolia Interior y Ningxia, mientras que la de la provincia de Shanxi fue más baja (Tabla 1). En cuanto a la prueba RT-PCR para el ARN BVDV, se encontraron 55 muestras positivas de todas las muestras de suero y la tasa positiva es del 8,14 %. En detalle, la tasa positiva (12,77%) fue más alta en Ningxia, seguida del 4% en Shanxi y del 2% en la provincia de Shandong, mientras que la tasa positiva (1%) en Mongolia Interior fue más baja (Tabla 1). Además, se identificaron 4 muestras de suero, que contenían anticuerpos para el ARN BVDV y BVDV, en las 376 muestras de suero de la provincia de Ningxia (Tabla 1).
 TABLA 1. La información sobre la muestra de suero objetivo que contiene el genoma BVDV y anticuerpos para BVDV.
TABLA 1. La información sobre la muestra de suero objetivo que contiene el genoma BVDV y anticuerpos para BVDV.
Ocho cepas BVDV aisladas de muestras de suero positiva
Cada muestra de suero que contenía ARN BVDV se utilizó para el aislamiento del virus. De las 55 muestras de suero positivas, se aislaron con éxito ocho cepas de BVDV. Según el ensayo de biotipo, hubo cuatro cepas BVDV con CPE (21NX-300, 21NX-53, 21SD-16 y 22NX-9) y cuatro cepas sin CPE (21NX-69, 21NM-44, 21SD-5 y 22NX-197) (Tabla 2). Para estimar el título viral de las cuatro cepas BVDV con CPE, el ensayo TCID50 mostró los diferentes títulos que oscilaban entre 105.0 y 107,0.
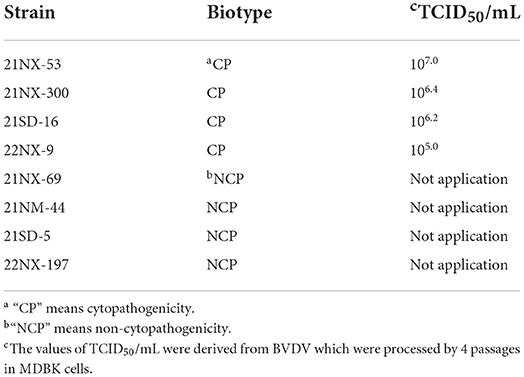 TABLA 2. La información sobre los ocho aislados BVDV presentados de muestras de suero.
TABLA 2. La información sobre los ocho aislados BVDV presentados de muestras de suero.
Las ocho cepas BVDV son el subgenotipo 1c
Dependiendo de los cebadores de PCR específicos, las ocho cepas BVDV se secuenciaron para todo el genoma. Se obtuvieron los 12 fragmentos superpuestos, que van de 931 a 1.168 nt de tamaño, (Tabla complementaria 1). Los ocho genomas virales oscilaron entre 12.132 y 12.453 de longitud, y su ORF osciló entre 11.691 y 11.727 nt de longitud, que codifica una poliproteína que oscilaba entre 3.896 aminoácidos y 3.989 aminoácidos de longitud (Tabla complementaria 3). Además, los 5’UTR y 3’UTR de estos aislados de BVDV oscilaron entre 282 y 317 nt y oscilaron entre 153 y 409 nt, respectivamente. Además, se utilizaron 28 cepas BVDV reportadas en China, que se secuenciaron para todo el genoma disponible en la base de datos GenBank, para aclarar la diversidad genética de las ocho cepas BVDV a nivel del genoma (Tabla complementaria 2). Un árbol filogenético que incluye representaciones de BVDV-1 con seis subgenotipos (1a-1d, 1m y 1q), BVDV-2 y BVDV-3 mostró que las ocho cepas de BVDV se agruparon en el mismo nodo terminal con las dos cepas del subgenotipo 1c (cepa GSTZ, acceso a GenBank no. Tensión MF172980.1 y GXNN1, número de acceso a GenBank. MT079816.1) (Figura 1). A pesar de las cepas GSTZ y GXNN1 que pertenecen a BVDV-1c, estas cepas BVDV tenían relaciones genéticas más estrechas con la cepa GSTZ que con GXNN1. Dado que la cepa GSTZ se aisló de la provincia de Gansu en el norte de China y la cepa GXNN1 se aisló de la provincia de Guangxi en el sur de China, el factor geográfico pareció participar en la vía evolutiva de las ocho cepas BVDV hasta cierto punto.
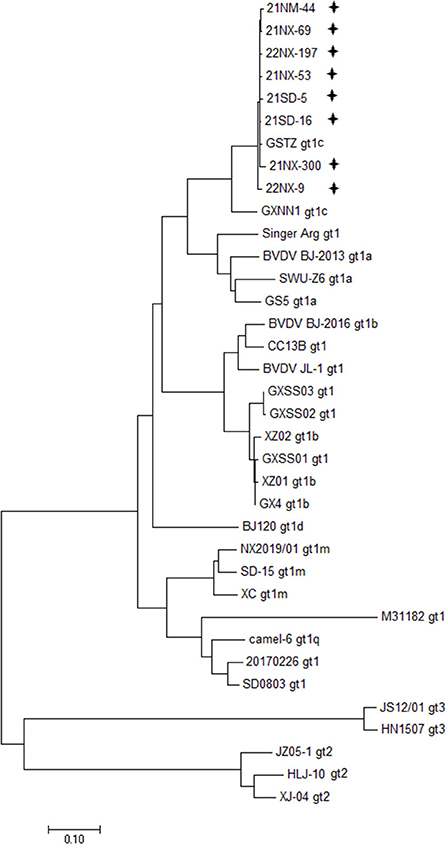 FIGURA 1. El análisis filogenético se basa en todo el genoma. Se creó un árbol filogenético utilizando los genomas completos de ocho aislados de BVDV y 28 cepas de referencia recuperadas de la base de datos GenBank. «+» de los aislados de BVDV identificados en las cuatro provincias de este estudio. Todos los aislados se agruparon en BVDV-1c.
FIGURA 1. El análisis filogenético se basa en todo el genoma. Se creó un árbol filogenético utilizando los genomas completos de ocho aislados de BVDV y 28 cepas de referencia recuperadas de la base de datos GenBank. «+» de los aislados de BVDV identificados en las cuatro provincias de este estudio. Todos los aislados se agruparon en BVDV-1c.
Fuertes fuerzas selectivas que suprimen el uso de codones para los aislados de BVDV
De acuerdo con los usos de nucleótidos de las ocho cepas (Tabla complementaria 4), el contenido de nucleótidos A fue más alto (valor promedio = 31,96 ± 0,05%), seguido de los de los nucleótidos G (26,09 ± 0,05%) y U (22,19 ± 0,04%), mientras que el contenido de nucleó Además, los nucleótidos A (valor promedio = 30,52 ± 0,08%) y G (26,81 ± 0,1 %) en la tercera posición de codón fueron similares con el contenido de nucleótidos correspondiente a nivel de codón, mientras que el contenido de nucleótidos U (20,22 ± 0,1 %) fue menor que el del nucleó Para investigar el papel de la restricción de la composición de nucleótidos en el sesgo de uso de codones sinónimos, se realizó el análisis de la RSCU para las ocho cepas BVDV. En general, las ocho cepas BVDV mostraban patrones de uso de codones sinónimos similares. Todos los codones sinónimos sobrerepresentados no siguieron el patrón de uso de codones con el extremo G/C o el extremo A/U, y todos los codones sinónimos que contenían dinucleótidos CpG fueron ligeramente seleccionados por las ocho cepas BVDV (Tabla complementaria 5). Aunque la restricción de composición de nucleótidos de BVDV ORF tuvo un fuerte efecto en el uso de codones sobrerrepresentados, incluidos AUA (Ile), UCA (Ser), CCA (Pro), AGA y AGG (Arg) y los infrarrepresentados (CUU, CUC para Leu, GUU para Val y CGU para Arg), Las características genéticas reflejaban las fuertes fuerzas selectivas que suprimían los usos sinónimos de codones para las ocho cepas BVDV. Curiosamente, las comparaciones de patrones de uso de codones sinónimos entre los aislados de BVDV con CPE y los que no tenían CPE mostraron los patrones de uso significativamente diferentes para la codificación UCG Ser (p = 0,017), la codificación CCC Pro (p = 0,004), la codificación GCA Ala (p = 0,03) y la codificación AAC Asn (p = 0,042).
Sobre la base del patrón general de uso de codones (datos de RSS) de los aislados de BVDV (Tabla complementaria 5), se realizó PCA para la relación genética entre las ocho cepas de BVDV y otras cepas reportadas en China. Para los 59 conjuntos de datos multidimensionales relevantes, PCA reduce la dimensionalidad de los datos para que se pueda realizar una visualización eficiente que captura la mayor parte de la variación. Aquí, los dos primeros ejes del análisis de PCA se utilizaron para establecer una visualización 2D de la diversidad genética para diferentes cepas BVDV en China. Después del análisis de PCA, la proyección de las cepas BVDV mostró que los ocho aislados de BVDV tenían las estrechas relaciones genéticas con la cepa GSTZ y mostraron obviamente diferencias con las demás (Figura 2). Además, las dos cepas de BVDV-3 tenían una diferencia obvia de otras y mostraban el uso de codones específico del genotipo, pero las tres cepas de BVDV-2 parecían mostrar una relación genética más estrecha con las tres cepas de BVDV-1 («20170226», «camel-6» y «SD0803») que otras cepas de BVDV-1 ( El resultado reflejó que las cepas BVDV-3, que surgieron en China, poseían una diferencia significativa con las de BVDV-1 y BVDV-2 en el uso de codón, y las cepas BVDV-1 y BVDV-2 no mostraron el uso de codón específico del genotipo. Además, los patrones generales de uso de codones para estas cepas de BVDV-1 obviamente mostraron una alta variante de uso de codones como sinónimo. Este resultado fue consistente con los informes anteriores para cepas BVDV-1 con altas variaciones de mutación (9, 32, 33).
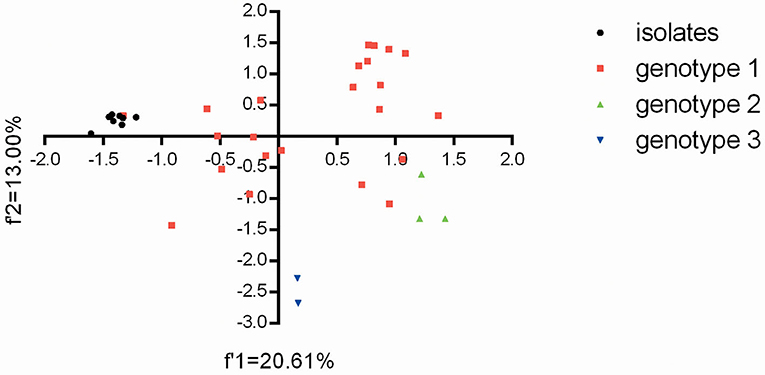 FIGURA 2. El gráfico para el uso general de codones se visualiza mediante el método PCA para estas cepas BVDV en este estudio.
FIGURA 2. El gráfico para el uso general de codones se visualiza mediante el método PCA para estas cepas BVDV en este estudio.
Discusión
BVDV es el más frecuente en la industria ganadera en China. El estudio anterior mostró que la prevalencia del ARN BVDV fue del 22,64 % en rebaños de ganado en China (10). Una revisión sistemática señaló que la prevalencia del ARN BVDV fue del 27,1 % en el ganado lechero de China (13). Sin embargo, se han realizado relativamente pocos estudios para la detección de ARN BVDV, la seroprevalencia viral, el aislamiento del virus, el genotipado y su distribución en terneros recién nacidos. La transmisión vertical de BVDV se produce cuando el virus se transmite desde la presa infectada a su descendencia. La infección fetal puede, dependiendo de la fase de gestación, provocar aborto, mortinato, efectos teratogénicos o, cuando la infección ocurre durante el primer trimestre, en el nacimiento de terneros inmunotolerantes, infectados persistentemente, virémicos (2, 3). Nuestro estudio ha ampliado la investigación anterior y ha mostrado la prevalencia serológica y de ARN para la infección por BVDV en terneros recién nacidos. Además, nuestro estudio demostró que la tasa positiva general de ARN BVDV fue del 8,14 % (55/676) y la prevalencia serológica fue del 9,6 % (66/676) en terneros recién nacidos en el norte de China. Actualmente, hay tres métodos principales de detección (RT-PCR, ELISA y aislamiento de virus) para la infección por BVDV en rebaños de ganado. Hasta cierto punto, las limitaciones en los muestreos y los métodos de detección podrían influir en la validez de la infección por BVDV en terneros recién nacidos. En general, la prevalencia de la infección por BVDV en terneros recién nacidos en el norte de China fue significativamente más débil que la infección general por BVDV en la población de ganado, lo que sugiere que la infección por BVDV en terneros recién nacidos a través de la transmisión vertical podría no servir como un factor importante de la epidemia de BVDV en los rebaños de ganado Dado que los terneros recién nacidos con infección por BVDV, especialmente los terneros recién nacidos de PI, representan una amenaza para la industria ganadera, estos terneros que desprenden enormes cantidades de BVDV a lo largo de sus vidas son la principal fuente para la propagación y perpetuación del virus dentro de las manadas de ganado individuales y para su transmisión a explotaciones no afectadas anteriormente (6 Por lo tanto, los terneros recién nacidos con infección por BVDV deben considerarse seriamente un objetivo importante para la erradicación de BVDV a través de la detección rápida o la estrategia de vacunación.
En comparación con RT-PCR para ARN BVDV y ELISA para antígenos / anticuerpos BVDV, el aislamiento del virus es el método estándar de detección de terneros recién nacidos infectados por BVDV. El ganado infectado por BVDV, especialmente el ganado PI, puede secretar grandes cantidades de BVDV en su suero (36, 37). Por lo tanto, el suero de la pantorrilla recién nacida es una muestra válida para el aislamiento de BVDV. En nuestro estudio, ocho cepas de BVDV (cuatro cepas de CP y cuatro de NCP) se aislaron con éxito del suero clínico de terneros recién nacidos. Estos aislados de BVDV con diferentes biotipos proporcionan más información biológica. Después del análisis de genotipado filogenético, los ocho aislados de BVDV se clasificaron en subgenotipo 1c y mostraron una estrecha relación genética con la cepa GSTZ que se aisló del yak en la provincia de Gansu (Figura 1). Un estudio anterior describió la circulación de al menos seis subgenotipos de BVDV-1 en China (1a, 1b, 1c, 1d 1m y 1q) con predominio de BVDV-1a y BVDV-1d (32, 37, 38). Actualmente, BVDV-1a y BVDV-1b siguen siendo los subgenotipos detectados con más frecuencia en la gran región, incluyendo Asia, Europa y América (39). Además, aunque las cepas BVDV-2 y BVDV-3 se han aislado en China (16, 19, 40, 41), nuestro estudio mostró que las dos especies de BVDV no parecen circular en terneros recién nacidos en el norte de China. BVDV-1c puede ser único para terneros recién nacidos en el norte de China y necesita ser monitoreado a partir de ahora. El estudio anterior señaló que BVDV-1a y BVDV-1c circulaban principalmente en la población ganadera (yak, ganado lechero y ganado vacuno) en China (37). De hecho, BVDV-1c es, con mucho, el que circula más comúnmente en Australia, y el ganado infectado por BVDV-1c sin signos respiratorios manifiestos, comúnmente mostró pirexia y leucopenia transitorias (42). En nuestro estudio, BVDV-1c desempeña un papel importante en la infección por BVDV de los terneros recién nacidos. La infección por BVDV-1c en terneros recién nacidos podría proporcionar una posible vía para las cepas de BVDV-1c en circulación en la población bovina.
Dado que BVDV es un virus ARN con alta tasa de mutación, los análisis de la frecuencia y el número de subgenotipos de BVDV en terneros recién nacidos son útiles para aclarar la tendencia evolutiva viral y las posibles fuentes infecciosas. En primer lugar, mostramos las características genéticas de los aislados de BVDV en terneros recién nacidos a nivel de uso de nucleótidos y sinónimos de codones. Aparte de la diversidad genética de las ocho cepas BVDV (Figura 1), los aislados de BVDV tenían patrones de uso de codones similares con la cepa GSTZ y un límite evolutivo obvio que se separaba de otros subgenotipos de BVDV-1, las cepas BVDV-2 y BVDV-3 que circulan en China (Figura 2), lo que sugiere además que la vía Las cepas aisladas de BVDV-1c con heterogeneidad del uso de codones podrían plantear preocupación relacionada con la aparición y propagación de nuevas variantes de BVDV en terneros recién nacidos, con posibles implicaciones para la salud animal y el control de enfermedades. De acuerdo con los patrones de uso de nucleótidos y codones sinónimos (Tablas complementarias 4, 5), la restricción de composición de nucleótidos y las fuerzas selectivas naturales cooperaron para impulsar el sesgo de uso de codones sinónimos de las ocho cepas BVDV. Además, en comparación de cada sesgo de uso de codones sinónimos entre los aislados de BVDV con biotipo de CP y los que tienen biotipo de NCP, se encontró interesante que cuatro codones sinónimos (UCG, CCC, GCA y AAC) mostraban las diferencias significativas en los patrones de uso. Esta característica genética sugiere que el sesgo de uso de codones sinónimos de BVDV podría participar en la configuración del biotipo BVDV. En general, el sesgo de uso de codones sinónimos puede participar en la mayoría de los aspectos del ciclo de vida viral. Este uso sesgado de codones sin sesgados no es neutral, sino que implica el sesgo de uso de nucleótidos (28, 29, 43), la estabilidad del ARNm (44, 45), la precisión y eficiencia de la traslación (46, 47) y la formación de plegamiento de proteínas (48). Cabe destacar que el BVDV no siguió el patrón de uso de nucleótidos de su huésped susceptible (es decir, ternero recién nacido) en el que el genoma de las células contiene un alto contenido de GC para realizar un ciclo de vida viral, pero acumuló contenido de adenina en su ORF (Tabla complementaria 4) y suprimió la selección de codones sinónimos con Se puede explicar que BVDV ORF tiende a suprimir los usos de codones sinónimos que contienen dinucleótidos de CpG para debilitar las respuestas inmunitarias del huésped infectado. Este fenómeno genético se puede explicar por las observaciones de que la selección natural derivada de la selección dependiente del huésped juega un papel importante en la eliminación de los dinucleótidos CG en el genoma viral (49, 50).
Conclusión
La distribución de los subgenotipos en terneros recién nacidos en el norte de China muestra una diferencia con respecto a otras regiones de China. A pesar de que BVDV-1a, 1b y 1c circulan principalmente en China, estos BVDV-1c parecen ser la principal fuente infecciosa de terneros recién nacidos en el norte de China, y las cepas aisladas de BVDV-1c muestran una tendencia evolutiva única de otras cepas de BVDV aisladas de China en el patrón de uso de nucle Se requieren más estudios para comprender completamente la variabilidad y la importancia del BVDV-1c. El monitoreo de estos aislados de BVDV que circulan en los terneros recién nacidos es un indicador útil con respecto al diseño de un programa de vacunación eficaz o una detección diagnóstica confiable.
Declaración de disponibilidad de datos
Los conjuntos de datos presentados en este estudio se pueden encontrar en repositorios en línea. Los nombres del repositorio/repositorios y el(los) número(s) de acceso se pueden encontrar en el artículo/materiales complementarios.
Contribuciones de los autores
HW, JZ y ZM: conceptualización. HW, MW, XF, XW, LZ, HL, XY, YL y DZ: metodología. YC y JL: software. HW, DZ, MW, XF, YL e YC: análisis formales. JZ y ZM: escritura: revisión y edición. XW, LZ, HL y XY: recolección de muestras de sueros. Todos los autores contribuyeron al artículo y aprobaron la versión presentada.
Financiación
El trabajo fue apoyado por los Fondos de Investigación Fundamental para las Universidades Centrales (n.o 31920220069) y los Fondos de Investigación Fundamental para la Introducción del Fondo de Investigación de Talento de la Universidad Northwest Minzu (No. xbmuyjrc202225).
Conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un posible conflicto de intereses.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o reclamo que pueda ser hecho por su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario de este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fvets.2022.984962/full#supplementary-material
Referencias
1. Smith DB, Meyers G, Bukh J, Gould EA, Monath T, Scott Muerhoff A, et al. Revisión propuesta de la taxonomía del género Pestivirus, familia Flaviviridae. J Gen Virol. (2017) 98: 21:06–12. doi: 10.1099/jgv.0.000873
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
2. Panadero C. Las manifestaciones clínicas de la diarrea viral bovina. Veterinario Clin North Am Food Anim Pract.(1995) 11:425-45. doi: 10.1016/S0749-0720(15)30460-6
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
3. Peterhans E, Schweizer M. Pestivirus: cómo sobremaniobrar a tus anfitriones. Veterinario Microbiol. (2010) 142:18-25. doi: 10.1016/j.vetmic.2009.09.038
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
4. Gunn GJ, Saatkamp HW, Humphry RW, Stott AW. Evaluar la presión económica y social para el control del virus de la diarrea viral bovina. Prev Vet Med. (2005) 72:149–62; discusión 215–9. doi: 10.1016/j.prevetmed.2005.08.012
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
5. Schweizer M, Peterhans E. Pestivirus. Annu Rev Anim Biosci. (2014) 2:141–63. doi: 10.1146/annurev-animal-022513-114209
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
6. Ezanno P, Fourichon C, Seegers H. Influencia de la estructura del rebaño y el tipo de introducción del virus en la propagación del virus de la diarrea viral bovina (BVDV) dentro de un rebaño lechero. Res del veterinario. (2008) 39:39. doi: 10.1051/vetres:2008016
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
7. Talafha AQ, Hirche SM, Ababneh MM, Al-Majali AM, Ababneh MM. Prevalencia y factores de riesgo asociados con la infección por el virus de la diarrea viral bovina en rebaños lecheros en Jordania. Trop Anim Health Prod.(2009) 41:499–506. doi: 10.1007/s11250-008-9214-6
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
8. Van Campen H. Epidemiología y control de la BVD en los EE. UU. Veterinario Microbiol. (2010) 142:94–8. doi: 10.1016/j.vetmic.2009.09.049
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
9. Giammarioli M, Ceglie L, Rossi E, Bazzucchi M, Casciari C, Petrini S, et al. Aumento de la diversidad genética de BVDV-1: hallazgos recientes e implicaciones de los mismos. Genes del virus. (2015) 50:147–51. doi: 10.1007/s11262-014-1132-2
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
10. Deng M, Ji S, Fei W, Raza S, He C, Chen Y, et al. Estudio de prevalencia y tipificación genética del virus de la diarrea viral bovina (BVDV) en cuatro especies bovinas en China. PLoS ONE. (2015) 1:e0121718. doi: 10.1371/journal.pone.0121718
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
11. Diao NC, Gong QL, Li JM, Zhao D, Li D, Zhao B, et al. Prevalencia del virus de la diarrea viral bovina (BVDV) en yaks entre 1987 y 2019 en China continental: una revisión sistemática y un metanálisis. Microb Pathog. (2020) 144:104185. doi: 10.1016/j.micpath.2020.104185
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
12. Wang L, Wu X, Wang C, Song C, Bao J, Du J. Origen y transmisión del virus de la diarrea viral bovina tipo 1 en China revelados por análisis filodinámico. Res Vet Sci. (2020) 128:162–9. doi: 10.1016/j.rvsc.2019.11.015
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
13. Ran X, Chen X, Ma L, Wen X, Zhai J, Wang M, et al. Una revisión sistemática y un metaanálisis de la epidemiología de la infección por el virus de la diarrea viral bovina (BVDV) en el ganado lechero en China. Acta Trop. (2019) 190:296-303. doi: 10.1016/j.actatropica.2018.08.031
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
14. Hou P, Zhao G, Wang H, He H. Prevalencia del virus de la diarrea viral bovina en rebaños de ganado lechero en el este de China. Trop Anim Health Prod. (2019) 51:791–8. doi: 10.1007/s11250-018-1751-z
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
15. Deng Y, Wang S, Liu R, Hao G. Diversidad genética de la infección por el virus de la diarrea viral bovina en cabras en el suroeste de China. J Vet Med. (2018) 2018:8274397. doi: 10.1155/2018/8274397
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
16. Shi H, Li H, Zhang Y, Yang L, Hu Y, Wang Z, et al. Diversidad genética de pestivirus bovinos detectados en granjas de ganado de patio trasero entre 2014 y 2019 en la provincia de Henan, China. Ciencia veterinaria frontal. (2020) 7:197. doi: 10.3389/fvets.2020.00197
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
17. Zhong F, Li N, Huang X, Guo Y, Chen H, Wang X, et al. tipificación genética y observación epidemiológica del virus de la diarrea viral bovina en el oeste de China. Genes del virus. (2011) 42:204–7. doi: 10.1007/s11262-010-0558-4
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
18. Deng Y, Sun CQ, Cao SJ, Lin T, Yuan SS, Zhang HB, et al. Alta prevalencia del virus de la diarrea viral bovina 1 en rebaños de cerdos chinos. Veterinario Microbiol. (2012) 159:490–493. doi: 10.1016/j.vetmic.2012.04.023
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
19. Zhu LQ, Lin YQ, Ding XY, Ren M, Tao J, Wang JY, et al. Secuenciación genómica y caracterización de un aislado chino del virus de la diarrea viral bovina 2. Acta Virol. (2009) 53:197–202. doi: 10.4149/av_2009_03_197
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
20. Wang W, Shi X, Chen C, Wu H. Caracterización genética de un virus de la diarrea viral bovina nocitopática 2b aislado de ganado en China. Genes del virus. (2014) 49:339–41. doi: 10.1007/s11262-014-1067-7
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
21. Zhang S, Tan B, Ding Y, Wang F, Guo L, Wen Y, et al. Secuencia completa del genoma y patogénesis del virus de la diarrea viral bovina JL-1 aislado del ganado en China. Virol J. (2014) 11:67. doi: 10.1186/1743-422X-11-67
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
22. Xie Z, Fan Q, Xie Z, Liu J, Pang Y, Deng X, et al. Secuencia completa del genoma de una cepa de virus de la diarrea viral bovina aislada en el sur de China. Genome Announc. (2014) 2:e00512–4. doi: 10.1128/genomeA.00512-14
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
23. Xue F, Zhu YM, Li J, Zhu LC, Ren XG, Feng JK, et al. Genotipo de virus de la diarrea viral bovina del ganado en China entre 2005 y 2008. Veterinario Microbiol. (2010) 143:379–83. doi: 10.1016/j.vetmic.2009.11.010
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
24. Zhou JH, Shang Y, Cao XA, Wang YN, Liu Y, Hu Y, et al. Posibles efectos de la infección por el virus de la hepatitis E en cerdos en la salud pública en China. Infect Genet Evol. (2019) 68:113–8. doi: 10.1016/j.meegid.2018.12.017
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
25. Tanton JB, Swanson B, Orozco E, Muñoz-Gutiérrez JF, Evermann JF, Ridpath JF. Las células microgliales de las ovejas inmortalizadas son permisivas para una amplia gama de virus de rumiantes. Veterinario Q. (2017) 37:52-6. doi: 10.1080/01652176.2017.1297550
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
26. Elikaei A, Sharifi Z, Hosseini SM, Latifi H, Musavi Hosseini MK. Inactivación de virus modelo suspendidos en plasma fresco congelado utilizando un nuevo dispositivo a base de azul de metileno. Irán J Microbiol. (2014) 6:41–5. doi: 10.1001/JAMA.289.8.959
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
27. Sharp PM, Li WH. El uso de codón en genes reguladores en Escherichia coli no refleja la selección de codones «raros». Ácidos nucleicos Res. (1986) 14:7737-49. doi: 10.1093/nar/14.19.7737
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
28. Ge Z, Li X, Cao X, Wang R, Hu W, Gen L, et al. Adaptación viral del fago estafilocócico: un análisis basado en el genoma de la preferencia selectiva basado en el sesgo de uso de codón. Genómica. (2020) 112:4657–65. doi: 10.1016/j.ygeno.2020.08.012
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
29. Li Y, Wang R, Wang H, Pu F, Feng X, Jin L, et al. Sesgo de uso de codones en el gen 13 relacionado con la autofagia en eucariotas: descubrimiento de la divergencia genética mediante la interacción entre los nucleotidos y los usos de codones. Las células frontales infectan el microbio. (2021) 11:771010. doi: 10.3389/fcimb.2021.771010
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
30. Zhou JH, Gao ZL, Zhang J, Ding YZ, Stipkovits L, Szathmary S, et al. El análisis del sesgo del codón del virus de la fiebre aftosa y la adaptación de este virus a los huéspedes. Infect Genet Evol. (2013) 14:105–10. doi: 10.1016/j.meegid.2012.09.020
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
31. Ben Salem K, Ben Abdelaziz A. Análisis de componentes principales (PCA). Túnez Med. (2021) 99:383–9. doi: 10.1186/s13040-022-00299-6
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
32. Deng M, Chen N, Guidarini C, Xu Z, Zhang J, Cai L, et al. Prevalencia y diversidad genética del virus de la diarrea viral bovina en rebaños lecheros de China. VetMicrobiol. (2020) 242:108565. doi: 10.1016/j.vetmic.2019.108565
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
33. Vilcek S, Durkovic B, Kolesarova M, Paton DJ. Diversidad genética de BVDV: consecuencias para la clasificación y la epidemiología molecular. Prev Vet Med. (2005) 72:31–5; discusión 215–9. doi: 10.1016/j.prevetmed.2005.08.004
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
34. Bitsch V, Hansen KE, Rønsholt L. Experiencias del programa danés para la erradicación de la diarrea por el virus bovino (BVD) 1994-1998 con especial referencia a la legislación y las causas de la infección. Veterinario Microbiol. (2000) 77:137–43. doi: 10.1016/S0378-1135(00)00270-4
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
35. Moennig V, Becher P. Control de la diarrea viral bovina. Patógenos. (2018) 7:29. doi: 10.3390/pathogens7010029
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
36. Lanyon SR, Hill FI, Reichel MP, Brownlie J. Diarrea viral bovina: patogénesis y diagnóstico. Veterinario J. (2014) 199:201–9. doi: 10.1016/j.tvjl.2013.07.024
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
37. Chang L, Qi Y, Liu D, Du Q, Zhao X, Tong D. Detección molecular y genotipado del virus de la diarrea viral bovina en el oeste de China. BMC Vet Res. (2021) 17:66. doi: 10.1186/s12917-021-02747-7
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
38. Gong X, Liu L, Zheng F, Chen Q, Li Z, Cao X, et al. Investigación molecular de la infección por el virus de la diarrea viral bovina en yaks (Bos gruniens) de Qinghai, China. Virol J. (2014) 11:29. doi: 10.1186/1743-422X-11-29
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
39. Yeşilbag K, Alpay G, Becher P. Variabilidad y distribución global de los subgenótipos del virus de la diarrea viral bovina. (2017) 9:128. doi: 10.3390/v9060128
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
40. Chen M, Liu M, Liu S, Shang Y. La infección por un pestivirus similar a HoBi provoca la muerte de los bovinos y a una enfermedad respiratoria grave en China. Transbound Emerg Dis. (2021) 68:1069–74. doi: 10.1111/tbed.13832
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
41. Mao L, Li W, Zhang W, Yang L, Jiang J. Secuencia del genoma de un nuevo pestivirus similar a Hobi en China. J Virol. (2012) 86:12444. doi: 10.1128/JVI.02159-12
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
42. Ambrose RK, Gravel JL, Commins MA, Fowler EV, Mahony TJ. Caracterización in vivo de cinco cepas del virus de la diarrea viral bovina 1 (Subgenetipo 1c). Patógenos. (2018) 7:12. doi: 10.3390/pathogens7010012
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
43. Han S, Hu W, Kan W, Ge Z, Song X, Li L, et al. Análisis de la genética y la patogénesis de Salmonella enterica QH con un espectro estrecho de resistencia a los antibióticos aislados del yak. Infect Genet Evol. (2020) 82:104293. doi: 10.1016/j.meegid.2020.104293
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
44. Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, et al. La optimización del codón es un determinante importante de la estabilidad del ARNm. Celda. (2015) 2160:1111-24. doi: 10.1016/j.cell.2015.02.029
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
45. Chen YH, Coller J. ¿Un código universal para la estabilidad del ARNm? Tendencias Genet. (2016) 32:687–8. doi: 10.1016/j.tig.2016.08.007
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
46. Hanson G, Coller J. Óptimo del codón, sesgo y uso en la traducción y la desintegración del ARNm. Nat Rev Mol Cell Biol. (2018) 19:20-30. doi: 10.1038/nrm.2017.91
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
47. Mauro VP, Chappell SA. Un análisis crítico de la optimización del codón en la terapia humana. Tendencias Mol Med. (2014) 20:604-13. doi: 10.1016/j.molmed.2014.09.003
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
48. Ling J, O’Donoghue P, Söll D. Flexibilidad del código genético en los microorganismos: mecanismos novedosos e impacto en la fisiología. Nat Rev Microbiol. (2015) 13:707-21. doi: 10.1038/nrmicro3568
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
49. Aragonès L, Guix S, Ribes E, Bosch A, Pintó RM. La selección de la cinética de traslación de ajuste fino como fuerza motriz del sesgo de uso de codones en la cápside del virus de la hepatitis A. PLoS Pathog. (2010) 6:e1000797. doi: 10.1371/journal.ppat.1000797
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
50. Greenbaum BD, Levine AJ, Bhanot G, Rabadan R. Patrones de evolución e imitación del gen huésped en la gripe y otros virus del ARN. PLoS Pathog. (2008) 4:e1000079. doi: 10.1371/journal.ppat.1000079
Resumen de PubMed | Texto completo de CrossRef | Google Scholar
Palabras clave: virus de la diarrea viral bovina, terneros recién nacidos, subgenotipo, codón sinónimo, dinucleótidos CpG
Cita: Wang H, Wang M, Feng X, Li Y, Zhang D, Cheng Y, Liu J, Wang X, Zhang L, La H, You X, Ma Z y Zhou J (2022) Características genéticas del virus de la diarrea viral bovina subgenotipo 1c en terneros recién nacidos en usos de nucleótidos y codones sinónimos. Frente. Veterinario. Sci. 9:984962. doi: 10.3389/fvets.2022.984962
Editado por:
Li Guangyu, Universidad Agrícola de Qingdao, China
Revisado por:
Wenjing Cui, Universidad de Jiangnan, China
Xin Cao, Universidad de Agricultura de Jilin, China
Feng Na, Academia China de Ciencias Agrícolas (CAAS), China
Copyright © 2022 Wang, Wang, Feng, Li, Zhang, Cheng, Liu, Wang, Zhang, La, You, Ma y Zhou. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia de Atribución de Creative Commons (CC BY). *Correspondencia: Jianhua Zhou, zhoujianhua@xbmu.edu.cn
†Estos autores han contribuido por igual a este trabajo
Descargo de responsabilidad: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o reclamación que pueda ser fabricado por su fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía