
Detección rápida de patógenos virales emergentes y de alta consecuencia en cerdos

Detección rápida de patógenos virales emergentes y de alta consecuencia en cerdos
 Alison C. Neujahr1
Alison C. Neujahr1  Duan S. Loy2
Duan S. Loy2  John Dustin Loy2
John Dustin Loy2  Bruce W. Brodersen2
Bruce W. Brodersen2  Samodha C. Fernando3,4,5*
Samodha C. Fernando3,4,5*- 1Departamento de Biosistemas Complejos, Universidad de Nebraska-Lincoln, Lincoln, NE, Estados Unidos
- 2Centro de Diagnóstico Veterinario de Nebraska, Universidad de Nebraska-Lincoln, Lincoln, NE, Estados Unidos
- 3Departamento de Ciencia Animal, Universidad de Nebraska-Lincoln, Lincoln, NE, Estados Unidos
- 4Departamento de Ciencias de los Alimentos, Universidad de Nebraska-Lincoln, Lincoln, NE, Estados Unidos
- 5Facultad de Ciencias Biológicas, Universidad de Nebraska-Lincoln, Lincoln, NE, Estados Unidos
Introducción: En la última década se ha observado una creciente aparición de nuevos patógenos animales. Los virus son uno de los principales contribuyentes al aumento de la aparición y, por lo tanto, la vigilancia veterinaria y los procedimientos de prueba son muy necesarios para detectar de forma rápida y precisa enfermedades animales de graves consecuencias, como la fiebre aftosa, la gripe aviar altamente patógena, la peste porcina clásica y la peste porcina africana. Los principales métodos de detección de estas enfermedades incluyen ensayos de PCR en tiempo real y anticuerpos específicos contra patógenos, entre otros. Sin embargo, debido a la deriva genética o al cambio en los genomas de los virus, la falta de detección de dichos patógenos es un riesgo con consecuencias devastadoras. Además, la aparición de nuevos patógenos sin conocimientos previos requiere métodos de detección no sesgados para su descubrimiento.
Métodos: Utilizando técnicas de enriquecimiento junto con la plataforma de secuenciación MinION™ de Oxford Nanopore Technologies, desarrollamos una línea de procesamiento y análisis de muestras para identificar virus de ADN y ARN y patógenos bacterianos a partir de muestras clínicas.
Resultados y discusión: La línea de procesamiento y análisis de muestras desarrollada permite la identificación simultánea de virus de ADN y ARN y patógenos bacterianos a partir de una sola muestra de tejido y proporciona resultados en menos de 12 h. La evaluación preliminar de este método utilizando virus sustitutos en diferentes matrices y utilizando muestras clínicas de animales con causalidad desconocida de la enfermedad, demostramos que este método se puede utilizar para detectar simultáneamente patógenos de múltiples dominios de vida simultáneamente con alta confianza.
1 Introducción
La industria porcina es una fuente importante de proteínas que representa un tercio del consumo de carne en todo el mundo (1, 2). Actualmente, se espera que la población mundial se duplique para 2050 (3) y que la producción de carne de cerdo aumente de 110,5 millones de toneladas métricas a 128,9 millones de toneladas métricas para el año 2031 (4). Como resultado, los productores han intensificado los sistemas de producción para satisfacer la demanda de los consumidores. Esta mayor intensificación ha dado lugar a altas densidades de animales, rápidas tasas de rotación de animales dentro de las salas de confinamiento y una mayor homogeneidad genética entre las instalaciones de producción porcina, lo que puede conducir a una mayor susceptibilidad a las enfermedades. Como resultado, se ha identificado una tasa creciente de patógenos porcinos y zoonóticos dentro de la industria (5). Dado que la carne de cerdo es un componente importante de la ganadería mundial, las consecuencias de las enfermedades infecciosas son impactantes (1). Un ejemplo de ello fue el reciente brote de peste porcina africana (PPA) en China, que dio lugar a la eutanasia masiva de más de 1 millón de animales (1). Por lo tanto, provocando un gran cambio económico en el mercado mundial de la carne de cerdo (1).
Para prevenir brotes, los organismos reguladores realizan actualmente pruebas de vigilancia veterinaria para detectar algunas enfermedades animales de graves consecuencias. Estas enfermedades incluyen el virus de la peste porcina africana (PPA), el virus de la peste porcina clásica (PPC), el virus de la pseudorrabia (PRV), el virus de la fiebre aftosa (FMDV) y el virus de la gripe A (IAV-S). Los programas de vigilancia suelen basarse en la detección mediante ensayos de reacción en cadena de la polimerasa (PCR) en tiempo real o con el uso de anticuerpos específicos junto con inmunoensayo ligado a enzimas (ELISA) (6). Por lo tanto, es necesaria información previa sobre la composición genética y/o los antígenos para la detección. Sin embargo, la deriva genética en los genomas de los patógenos puede impedir la detección, ya que los ensayos son muy específicos de la secuencia genética. Por ejemplo, virus como el de la influenza poseen genomas segmentados. Por lo tanto, estos virus pueden cambiar rápidamente sus antígenos, virulencia y capacidad para replicarse en las especies huésped a través del cambio genético (7). Como resultado, estos virus pueden pasar desapercibidos en los ensayos que utilizan regiones específicas para la detección. Además, este enfoque evita la identificación o detección de patógenos nuevos o emergentes. Con la aparición de nuevos patógenos, se necesita un conjunto alternativo de herramientas y métodos de vigilancia que puedan monitorear simultáneamente patógenos conocidos y desconocidos.
A través del desarrollo de métodos rápidos de secuenciación en el campo (8), la secuenciación es una herramienta atractiva para el diagnóstico y la vigilancia de patógenos (8). La tecnología Oxford Nanopore Technology (ONT) ofrece nuevas oportunidades para la detección y vigilancia de patógenos nuevos y emergentes. En 2014, ONT lanzó una plataforma llamada MinION,™ un secuenciador portátil con un costo de secuenciación reducido y salida de datos en tiempo real (8). La secuenciación de nanoporos difiere de otros secuenciadores, ya que funciona midiendo la corriente iónica a medida que un nucleótido pasa a través de un poro (9). Dentro del MinION,™ el rendimiento típico de los datos de secuenciación es de 10 a 20 Gb con un tiempo máximo de secuenciación de 72 h (9). La reducción en el costo de secuenciación, el aumento de la longitud de la secuenciación, la rápida respuesta y la facilidad de uso del instrumento han convertido a esta tecnología en una herramienta de diagnóstico atractiva en el campo para la atención y vigilancia del paciente (8). Además, la secuenciación de lectura larga de la ONT permite llenar los vacíos dentro de los genomas de la secuenciación de lectura corta (8), lo que permite un conocimiento más profundo de los genomas incompletos. En este estudio, investigamos el potencial de la secuenciación en tiempo real, utilizando la plataforma ONT MinION™, para identificar y monitorear patógenos ganaderos para desarrollar una herramienta de vigilancia y diagnóstico de patógenos animales emergentes utilizando cerdos como modelo. En este estudio, se utilizaron virus de la diarrea viral bovina (BVD), herpesvirus bovino-1 (IBR) y virus porcino del Valle de Seneca A (SVV) como virus sustitutos en lugar del virus de la peste porcina clásica (PPC), el virus de la pseudorrabia (PRV) y el virus de la fiebre aftosa (FMDV), respectivamente. Se utilizó directamente el virus de la influenza A (IAV-S) en lugar de un virus sustituto. Además, en lugar del virus de la peste porcina africana, se utilizó ADN sintético (gBlocks).
2 Métodos
2.1 Crecimiento de virus sustitutos
Se inoculó una monocapa de suero confluente al 75% de cornetes nasales adaptados para caballos (BT CRL-1390), sobre células de cornetes bovinos (BT) y testículos porcinos (ST), con 6 ×103 TCID50/mL BVD, IBR y SVV, respectivamente, en un2 matraz de cultivo de tejidos. Las células infectadas se cultivaron en medios esenciales mínimos (MEM; GIBCO, Grand Island, NY) al 5% de CO2, 37°C durante 1 h. Durante la incubación, los matraces se agitaron suavemente cada 15 minutos para garantizar una distribución uniforme del virus. Después de 1 h de incubación, se añadieron medios esenciales mínimos a los matraces para elevar el volumen hasta 30 ml. Los matraces se mantuvieron al 5% de CO2, 37 °C durante 3 días, para todos los virus, y se observó el efecto citopático (CPE). Una vez que el CPE alcanzó aproximadamente el 50 al 70% del total de las celdas, los matraces se colocaron en un congelador de -80 °C durante 30 min. Los matraces se descongelaron y el contenido se transfirió a tubos de centrífuga de 50 ml. Los tubos se centrifugaron a 2.000 rpm durante 5 min y el sobrenadante (stock de virus) se transfirió a un nuevo tubo de centrífuga de 50 mL. En total, se prepararon alícuotas de 1 mL de stock de virus en tubos criogénicos de 2 mL y se almacenaron a -80 °C hasta que se utilizaron como virus sustitutos para la aparición de picos tisulares. El número de copias virales se estimó utilizando valores de Ct de PCR en tiempo real, como se describió anteriormente, junto con la evaluación de la concentración de partículas del virus utilizando TCID50/mL viral (10-17). La información sobre el número estimado de copias del virus para cada cultivo de virus puro se puede encontrar en la Tabla Suplementaria S1. En el caso de la peste porcina africana (PPA), se sintetizaron bloques g a partir de las secuencias genómicas de la PPA para regiones seleccionadas (Tabla suplementaria S2) y se utilizaron como sustitutos del virus de la PPA. La información sobre la secuencia de los genes de la PPA se obtuvo de la base de datos del NCBI (NCBI:txid10497).
2.2 Identificación de virus sustitutos a partir de diferentes matrices y muestras desconocidas
Para evaluar la aplicabilidad del uso de estrategias de secuenciación en tiempo real para detectar e identificar patógenos, incluidos patógenos virales y bacterianos, los patógenos virales se mezclaron con muestras de tejido a 10 mL de cultivo de virus por 25 mL de homogeneizado de tejido (utilizando tejido pulmonar bovino). Se utilizó tejido pulmonar bovino como matrices para la detección de picos de BVD e IBR. Las muestras resultantes se sometieron a la extracción total de ácidos nucleicos como se describe a continuación. En resumen, las muestras de tejido que contenían las partículas del virus se molieron utilizando un disruptor de muestras (TissueLyser II, Qiagen, Hilden, Alemania) durante 2 minutos a 18 Hz y se filtraron a través de filtros de 0,2 μm (Thermo Scientific, Waltman MA, Estados Unidos) para eliminar cualquier contaminación de células huésped o bacterianas. Después de la filtración, se utilizó la ultracentrifugación para granular partículas de virus a 13.000 × g durante 1 h. Las muestras concentradas de virus se resuspendieron en 30 L de agua libre de nucleasas y se trataron con DNAseI (Thermo Scientific, Waltman MA, Estados Unidos) y RNAseA (Thermo Scientific, Waltman MA, Estados Unidos) para eliminar cualquier ADN y ARN que flotara libremente antes de someterlos a la extracción total de ácidos nucleicos utilizando el kit de ARN/ADN de patógenos MagMAX (Applied Biosystems, Waltman MA, Estados Unidos), extracción de alto volumen de acuerdo con el protocolo del fabricante.
Además, para validar aún más el método basado en secuenciación de lectura larga desarrollado en este estudio para su utilidad en muestras clínicas, este enfoque se evaluó utilizando muestras clínicas obtenidas del Centro de Diagnóstico Veterinario de la Universidad de Nebraska-Lincoln. El estudio fue ciego al personal de laboratorio que realizaba los experimentos en busca de presuntos patógenos virales y bacterianos en la muestra. Las muestras habían sido analizadas previamente y el agente causal se identificó para las muestras mediante análisis de PCR en tiempo real. Las muestras de tejido, ciegas para nosotros, que contenían patógenos «desconocidos» de origen bovino y porcino se filtraron a través de un filtro de 0,8 μm en lugar del filtro de 0,2 μm para eliminar las células huésped y retener bacterias y virus, y se sometieron a un tratamiento con ADNasa y ARNasa y extracción de ácidos nucleicos como se describió anteriormente. Se realizó un cambio en el tamaño del filtro de 0,2 μm a 0,8 μm para muestras «desconocidas» para recuperar patógenos bacterianos y virales de la misma muestra.
2.3 Experimentos con gBlock de peste porcina africana
Las muestras de amígdalas bovinas se extrajeron utilizando el protocolo descrito anteriormente y los fragmentos de ADN gblock de la PPA se agruparon en una proporción de 1:1:1 y se agregaron al ácido nucleico extraído de las muestras en la cantidad conocida de 224 y 2.242 copias de fragmentos de genes de PPA antes de la preparación de la biblioteca. Los números exactos de copias para cada conjunto de gBlock de PPA se pueden encontrar en la Tabla Suplementaria S2.
2.4 Síntesis de ADNc para la detección de virus de ARN
Los ácidos nucleicos totales extraídos se analizaron utilizando chips de ADN de alta sensibilidad y chips de ARN procariota Pico utilizando un Agilent BioAnalyzer 2000 (Agilent Technologies, Santa Clara, CA, Estados Unidos). El ARN presente en la muestra se convirtió en ADNc utilizando el kit de síntesis de ADNc de primera cadena ProtoScript II (NEBNext Biosciences, Ipswich MA, Estados Unidos) y el módulo de síntesis de segunda cadena de ARN no direccional NEBNext Ultra II (NEBNext Biosciences, Ipswich MA, Estados Unidos), de acuerdo con el protocolo del fabricante con la excepción del uso de pentadecámeros aleatorios para aumentar el rendimiento viral de ADNc como se describió anteriormente por Stangegaard et al. (18). Como medida adicional de control de calidad para garantizar que las partículas virales no se perdieran durante la síntesis de ADNc, se utilizaron cebadores específicos de cepas de virus para BVD, SVV, IBR e IAV-S para amplificar los virus objetivo (Tabla suplementaria S3). Los cebadores se muestran en la Tabla Suplementaria S3 y se obtuvieron de informes de literatura previa (11, 14, 15, 17). Las muestras resultantes, que contenían ADN y ADNc, se cuantificaron mediante el ensayo de fluorescencia de alta sensibilidad Denovix (Denovix Inc., Wilmington DE, Estados Unidos) y se utilizaron para la preparación de bibliotecas para la secuenciación en la plataforma de secuenciación de nanoporos.
2.5 Preparación de la biblioteca de nanoporos
La mezcla de ácidos nucleicos que contenía ADNc y ADN se utilizó para la preparación de la biblioteca utilizando el kit de código de barras de PCR SQK-P004 (Oxford Nanopore Technologies, Oxford, Reino Unido) con las dos modificaciones siguientes: el tiempo de ligadura se amplió de 10 min a 30 min y los ciclos de PCR se incrementaron de 15 ciclos a 18-20 ciclos. Después de la preparación de la biblioteca, las muestras se evaluaron utilizando el chip de ADN de alta sensibilidad Agilent BioAnalyzer 2000 (Agilent Technologies, Santa Clara, CA, Estados Unidos) para visualizar las bibliotecas y evaluar la calidad y el tamaño de las bibliotecas preparadas. Las concentraciones de la biblioteca se evaluaron mediante un ensayo de fluorescencia de alta sensibilidad de Denovix (Denovix Inc., Wilmington DE, Estados Unidos). Las concentraciones de la biblioteca se ajustaron a 0,88 nM y se agruparon y secuenciaron en la celda de flujo SpotON de Oxford Nanopore Technologies 9.4 o 10.3 (Oxford Nanopore Technologies, Oxford, Reino Unido) de acuerdo con el protocolo del fabricante. La secuenciación se realizó durante 72 h y el umbral de calidad de lectura se estableció en un Q-Score de >7. Los archivos Fastq se generaron dentro del software MinKNOW y se configuraron para generar 4.000 lecturas por archivo fastq. El flujo de trabajo completo se puede ver en la Figura 1.
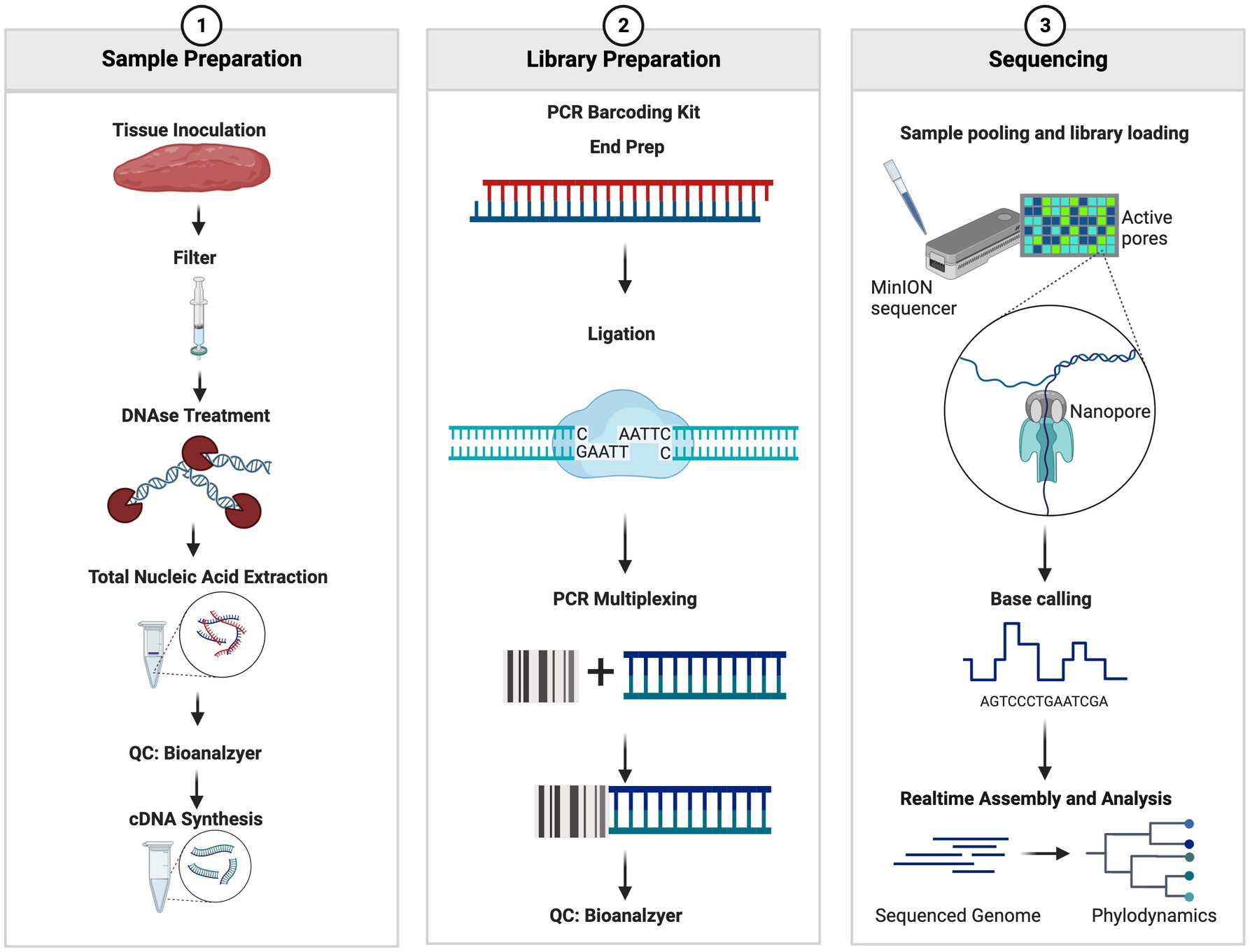 Figura 1. Flujo de trabajo resumido de la preparación de la muestra. La cifra se generó en biorender.com.
Figura 1. Flujo de trabajo resumido de la preparación de la muestra. La cifra se generó en biorender.com.
2.6 Análisis de datos
Los archivos fastq sin procesar se clasificaron con códigos de barras y se concatenaron en un solo archivo usando el comando «cat *fastq > > nombre de archivo. Fastq.» Se utilizó la herramienta informática Porechop (19) para eliminar los adaptadores asociados a cada archivo fastq. Después del recorte del adaptador, los archivos fastq se alinearon con una base de datos personalizada utilizando Centrifuge (20). La base de datos personalizada incluía todos los genomas conocidos de arqueas, bacterias, virus, ganado y cerdos presentes en la base de datos RefSeq del NCBI. La base de datos de referencia se creó utilizando Centrifuge (20). Los resultados del informe K del paquete Centrifuge se visualizan utilizando Pavian (21).
Los datos se analizaron en incrementos de 4.000 lecturas, mientras se realizaba la secuenciación para identificar una profundidad de secuenciación suficiente. Cada conjunto de archivos fastq generados se concatenó, recortó y alineó en una base de datos personalizada como se describió anteriormente. A medida que los archivos se generaban en conjuntos de 4.000 lecturas y se analizaban, se hizo un gráfico de líneas para identificar cuántos números de lecturas generadas creaban una meseta, lo que significaba que se había alcanzado la profundidad de lectura y no era necesaria una secuenciación adicional de cada muestra.
2.7 Disponibilidad de datos y materiales
La información detallada sobre el pipeline informático se puede encontrar en la página de Github de Fernando Lab.1 El conjunto de datos generado y analizado se puede encontrar en el número de acceso PRJNA1045613 de BioProject del archivo de lectura de secuencia (SRA) del Centro Nacional de Información Biotecnológica (NCBI).
2.8 Identificación de la profundidad de lectura para la identificación y validación de especies mediante PCR en tiempo real
Para estimar la profundidad de lectura mínima requerida para identificar patógenos con confianza en una muestra determinada, realizamos un análisis incremental de lecturas utilizando incrementos de 4.000 lecturas. El análisis bioinformático se realizó como se indicó anteriormente utilizando conjuntos de datos incrementales de 4.000, 8.000, 12.000 y 16.000 lecturas y se monitoreó la distribución taxonómica y de patógenos para cada conjunto de datos para identificar umbrales de lectura.
Además del análisis de lectura en profundidad, se realizó un análisis convencional de PCR en tiempo real para determinar la presencia viral descrita anteriormente con modificaciones menores (11, 14, 17, 22, 23). Brevemente, los ensayos de PCR en tiempo real se modificaron ligeramente y optimizaron para utilizar una mezcla maestra comercial Reliance One-Step Multiplex Super Mix (Bio-Rad Laboratories, Inc) o TaqMan™ Fast Virus 1-Step Master Mix (Thermo Fisher Scientific), así como un sistema de control interno adicional en la Biorad CFX96 Real-time PCR (Bio-Rad Laboratories, Inc.) o el sistema de PCR en tiempo real ABI 7500 FAST (Thermo Fisher Scientific). Todos los ensayos se realizaron como parte de protocolos aprobados bajo el sistema de calidad del Centro de Diagnóstico Veterinario de Nebraska que han sido validados para su uso.
3 Resultados
3.1 Posible uso de una nueva herramienta de diagnóstico y vigilancia de virus basada en secuencias
Los virus sustitutos se propagaron utilizando sistemas de cultivo celular y se utilizaron para desarrollar y validar un nuevo protocolo para la identificación de patógenos virales y bacterianos conocidos y desconocidos a partir de una sola muestra. Con este fin, se analizaron muestras de control enriquecidas con virus ssRNA y dsDNA de sentido negativo y positivo, tanto individualmente como en combinación, para evaluar la aplicabilidad del método desarrollado para identificar simultáneamente múltiples virus patógenos. La detección de virus individuales se probó contra virus de ADN y ARN, que incluían dos virus sustitutos, el SVV porcino y el BVD, para validar que el protocolo podría usarse ampliamente para identificar virus de ADN y ARN dentro de una muestra. Los sobrenadantes de cultivo celular de SVV (virus ARN) cuando se inocularon en matrices de tejido porcino permitieron la identificación del SVV con el 19,82% del total de lecturas (Figura 2A), representando el virus objetivo incluso con un nivel muy alto de contaminación del ADN eucariota. Además, una muestra enriquecida con BVD (virus ARN) en el tejido pulmonar bovino cuando se analizó utilizando el enfoque descrito en este estudio identificó que el 20,89% de las lecturas pertenecían a origen viral. Además, el 51,8% de las lecturas pertenecían a Mycoplasma (Figura 2B).
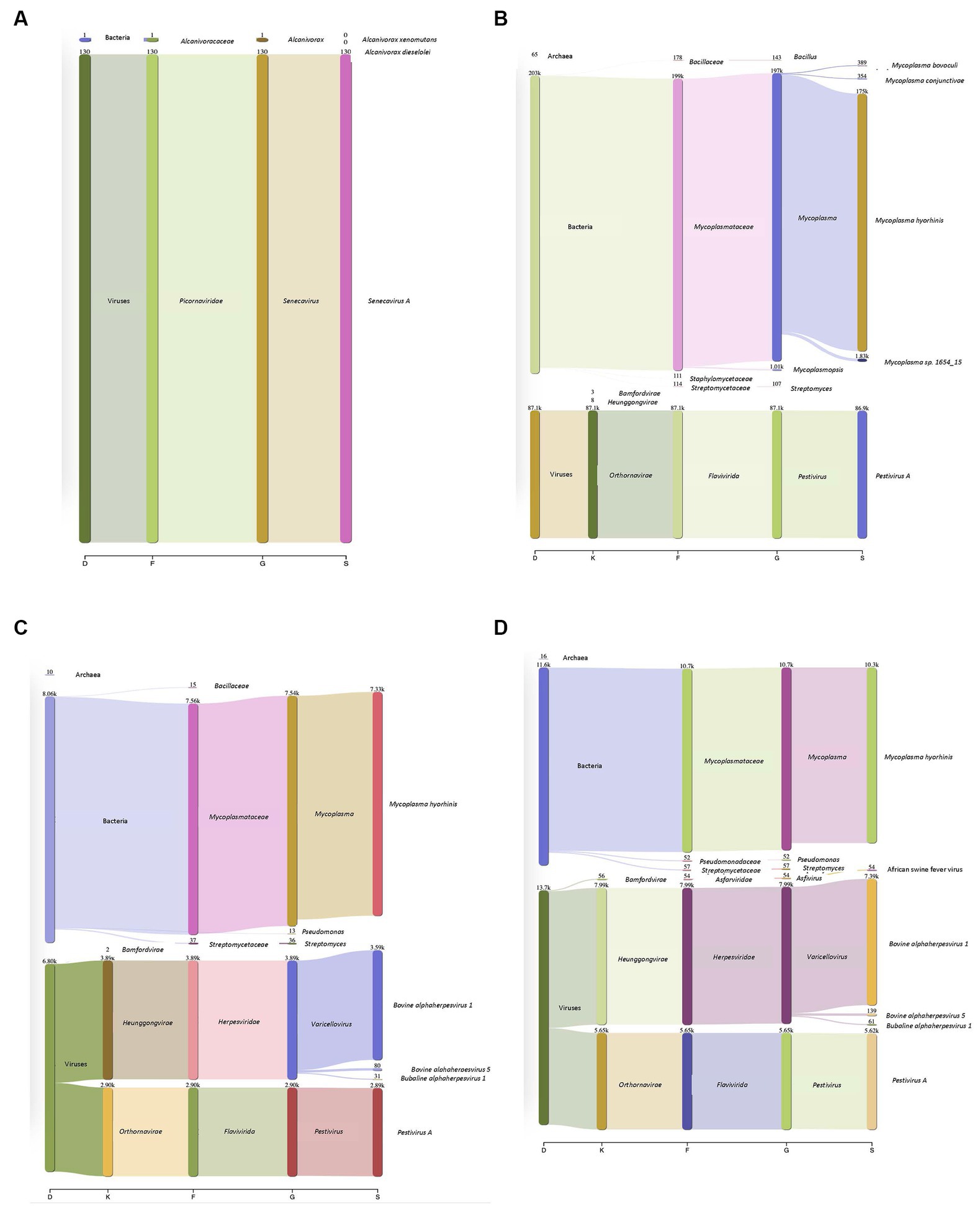 Figura 2. Detección de virus de ADN y ARN en diferentes matrices. (A) Virus porcino del Valle de Séneca (SVV) identificado en una muestra de tejido de amígdala porcina con púas. D-Dominio, F-familia, G-género y S-especie. Los números indican cuántas lecturas se anotaron para cada dominio, familia, género y especie. (B) El virus de la diarrea viral bovina (BVD) se introdujo en una muestra de tejido pulmonar bovino y se recuperaron 86,000 lecturas que pertenecían a BVD. Además, se identificó micoplasma dentro de esta muestra a pesar de filtrar a través de un filtro de 0,2 μm. (C) La complejidad del protocolo se incrementó al aumentar el número de virus dentro de una muestra dada (tanto ADN como ARN). Los resultados mostraron que tanto el ADN como el ARN pueden ser detectados. Las partículas virales se introdujeron en una muestra de tejido pulmonar bovino. (D) Se añadieron bloques g de peste porcina africana (PPA) a una muestra compleja que consistía tanto en alfaherpesvirus bovino (IBR, virus ADN) como en Pestivirus A (BVD, un virus ARN) que se introdujeron en tejido pulmonar bovino. Se detectaron los tres tipos de virus.
Figura 2. Detección de virus de ADN y ARN en diferentes matrices. (A) Virus porcino del Valle de Séneca (SVV) identificado en una muestra de tejido de amígdala porcina con púas. D-Dominio, F-familia, G-género y S-especie. Los números indican cuántas lecturas se anotaron para cada dominio, familia, género y especie. (B) El virus de la diarrea viral bovina (BVD) se introdujo en una muestra de tejido pulmonar bovino y se recuperaron 86,000 lecturas que pertenecían a BVD. Además, se identificó micoplasma dentro de esta muestra a pesar de filtrar a través de un filtro de 0,2 μm. (C) La complejidad del protocolo se incrementó al aumentar el número de virus dentro de una muestra dada (tanto ADN como ARN). Los resultados mostraron que tanto el ADN como el ARN pueden ser detectados. Las partículas virales se introdujeron en una muestra de tejido pulmonar bovino. (D) Se añadieron bloques g de peste porcina africana (PPA) a una muestra compleja que consistía tanto en alfaherpesvirus bovino (IBR, virus ADN) como en Pestivirus A (BVD, un virus ARN) que se introdujeron en tejido pulmonar bovino. Se detectaron los tres tipos de virus.
Además de los virus individuales introducidos en muestras de tejido, se inocularon combinaciones de virus de ADN y ARN en tejidos pulmonares bovinos para evaluar la detección de múltiples virus dentro de una muestra determinada. Las mezclas virales identificaron con éxito todas las combinaciones virales inoculadas. Cuando se analizó una muestra que contenía tanto un virus de ADN (Herpesvirus bovino-1 (IBR)) como de ARN (BVD) a través del protocolo desarrollado de preparación, secuenciación y análisis de muestras, el 19,2% de las lecturas se identificaron como de origen viral. Tanto las lecturas virales de IBR como de BVD se detectaron en la muestra con un 10,96 y un 8,2%, respectivamente (Figura 2C). Del mismo modo, cuando la complejidad se incrementó aún más al incluir los bloques g de la peste porcina africana (PPA) en 224 copias o 2.242 copias de ADN sintético en el tejido pulmonar que contenía IBR y BVD, el 26,6% de las lecturas se identificaron como lecturas virales en la muestra que consistía en IBR:BVD:ASF2242 (Figura 2D). Cuando solo se incluyeron 224 copias genéticas de PPA dentro de una matriz de muestra, solo se identificó 1 lectura como perteneciente a PPA. Sin embargo, con 2.242 copias de PPA, se identificaron 54 lecturas pertenecientes a la PPA, lo que representó el 0,11% del total de lecturas dentro de la matriz de muestra dada (IBR:BVD:ASF2242). En la Tabla 1 se puede encontrar una tabla de la matriz de muestra, el número de copias y los porcentajes de lectura.
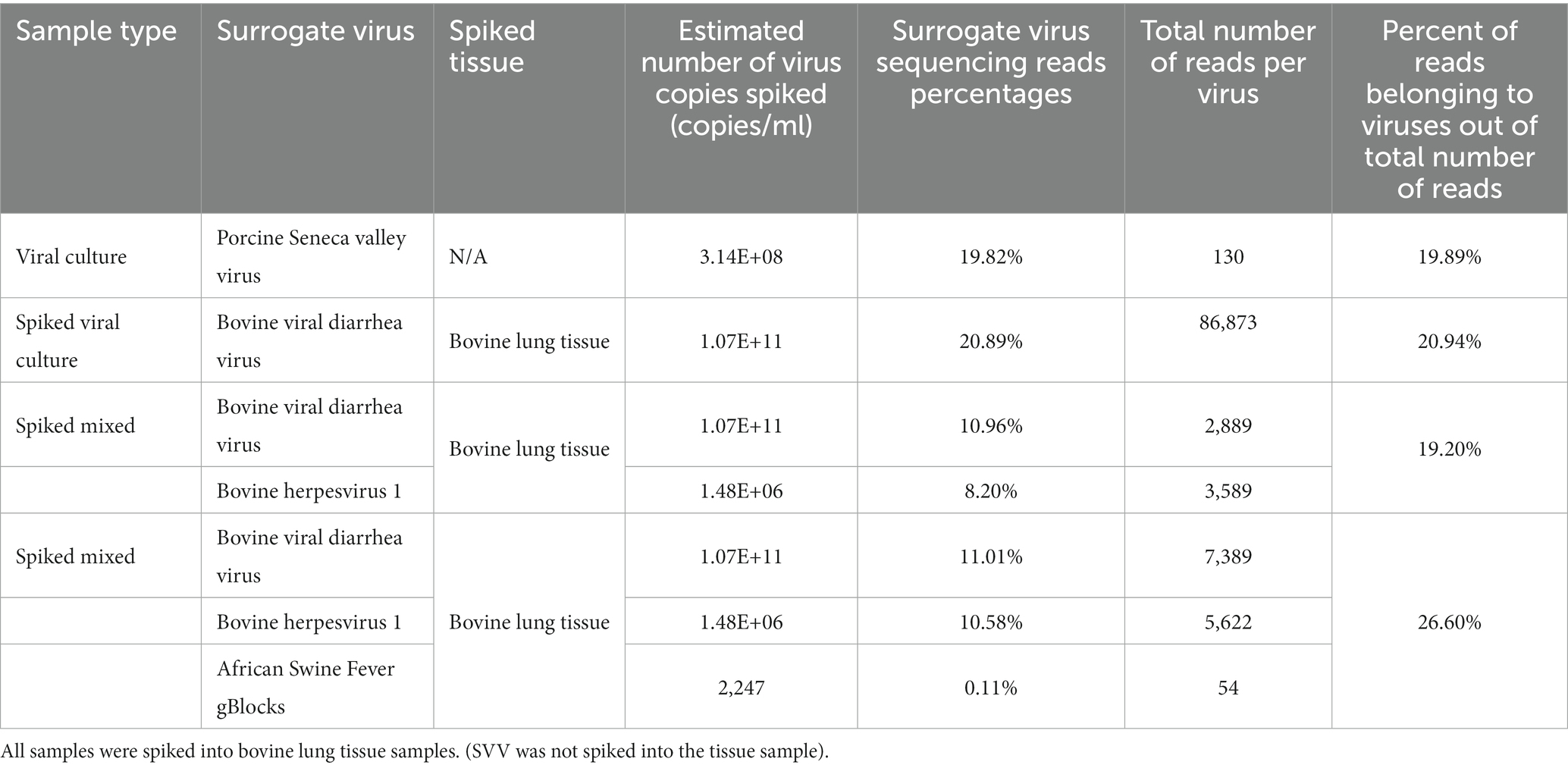 Tabla 1. Matriz de muestra, número de copias/ml y porcentaje de lectura identificados dentro de una matriz dada.
Tabla 1. Matriz de muestra, número de copias/ml y porcentaje de lectura identificados dentro de una matriz dada.
3.2 Identificación simultánea de patógenos virales y bacterianos en muestras clínicas
Para validar aún más nuestro protocolo desarrollado y evaluar la aplicabilidad de la técnica desarrollada para muestras clínicas, evaluamos dos muestras de cerdos y una muestra de bovino que eran «desconocidas» o ciegas para nosotros utilizando el protocolo descrito anteriormente. Además de las muestras que no se nos han dado a cuenta, comparamos los resultados encontrados en nuestro protocolo de secuenciación con la RT-PCR convencional. Se identificó que las dos muestras porcinas desconocidas contenían Circovirus porcino 2 (PCV2), virus del síndrome respiratorio y reproductivo porcino (PRRSV) y SVV porcino y fueron consistentes con los resultados obtenidos a través de RT-PCR convencional (Tabla Suplementaria S4). Se identificó que la muestra bovina contenía los patógenos Mycoplasma bovis, Mannheimia haemolytica, Pasteurellaceae y BVD y fue consistente con los resultados de la PCR en tiempo real (Tabla suplementaria S4). Como se muestra en la Tabla Suplementaria S4, se realizó RT-PCR para muestras desconocidas antes de la preparación de la biblioteca de ONT para obtener una abundancia viral y bacteriana. Además, después de la preparación de la biblioteca, se realizó una RT-PCR para identificar los valores de abundancia de patógenos después de la preparación de la biblioteca (Tabla Suplementaria S4).
3.3 Identificación de la profundidad de lectura mínima para detectar con precisión virus y bacterias patógenas
Para evaluar la profundidad de lectura a la que se pueden detectar con precisión los patógenos virales y bacterianos, y para reducir el costo y el tiempo de secuenciación, configuramos la secuencia para generar lecturas de secuencia en conjuntos de 4,000 lecturas y analizamos los datos en orden secuencial a medida que salían de la secuencia para evaluar a qué profundidad de lectura se estabilizaría la detección de patógenos y proporcionaría resultados consistentes en la identificación de patógenos. Para ello, evaluamos series de secuenciación que contenían diferentes combinaciones de virus y bacterias, además de las muestras clínicas desconocidas. Este enfoque se utilizó en combinaciones virales que contenían ADN y patógenos virales y patógenos bacterianos de ssRNA de sentido positivo y negativo. Se identificó una profundidad de lectura suficiente cuando la cobertura de virus con picos conocidos alcanzó una meseta en el porcentaje de lecturas totales. Con este fin, evaluamos la abundancia de lecturas de patógenos como una proporción de las lecturas totales, ya que se generó a partir de la secuenciación en incrementos de 4.000 lecturas. Este análisis identificó que a las 12.000 lecturas, podíamos identificar de forma consistente y precisa los patógenos presentes en una muestra (Figura 3) en función del número de copias iniciales. Por lo tanto, si se ha alcanzado una profundidad de lectura suficiente, la ejecución de secuenciación podría detenerse y la celda de flujo podría lavarse para un uso adicional de secuenciación, lo que reduciría los costos y ahorraría tiempo.
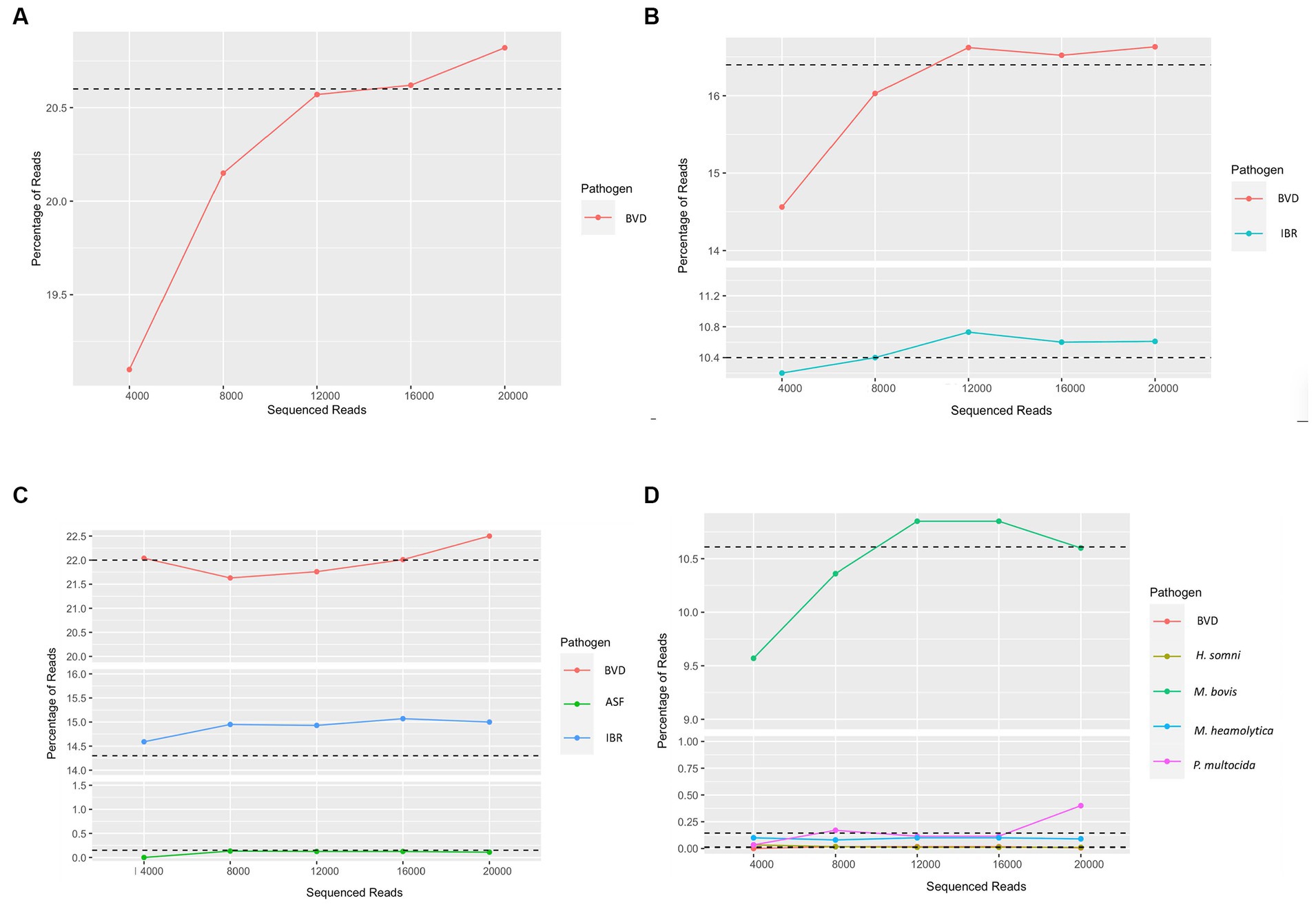 Figura 3. Determinación de la profundidad de lectura necesaria para una evaluación segura de la muestra. (A) Detección del virus de la diarrea viral bovina (BVD), (B) Detección del virus de la diarrea viral bovina (BVD) y alfaherpesvirus bovino (IBR), (C) Detección del virus de la diarrea viral bovina (BVD), peste porcina africana (PPA) y alfaherpesvirus bovino (IBR), (D) Muestra bovina desconocida utilizada para identificar patógenos potenciales. Las salidas son del primero al quinto archivo fastq generado. Se realizaron corridas de secuenciación hasta agotar todos los poros para estimar la profundidad de secuenciación. La dependencia de la muestra, 12.000 lecturas o los tres primeros archivos fastq mostraron una profundidad adecuada para el diagnóstico clínico de las muestras de un patógeno determinado.
Figura 3. Determinación de la profundidad de lectura necesaria para una evaluación segura de la muestra. (A) Detección del virus de la diarrea viral bovina (BVD), (B) Detección del virus de la diarrea viral bovina (BVD) y alfaherpesvirus bovino (IBR), (C) Detección del virus de la diarrea viral bovina (BVD), peste porcina africana (PPA) y alfaherpesvirus bovino (IBR), (D) Muestra bovina desconocida utilizada para identificar patógenos potenciales. Las salidas son del primero al quinto archivo fastq generado. Se realizaron corridas de secuenciación hasta agotar todos los poros para estimar la profundidad de secuenciación. La dependencia de la muestra, 12.000 lecturas o los tres primeros archivos fastq mostraron una profundidad adecuada para el diagnóstico clínico de las muestras de un patógeno determinado.
4 Discusión
La necesidad de una rápida identificación de patógenos nuevos y emergentes se ha convertido en un requisito para garantizar la detección oportuna y precisa de patógenos para facilitar la respuesta y las intervenciones. En virología veterinaria, la secuenciación de nueva generación (NGS) se ha utilizado para probar matrices para la caracterización e identificación de patógenos virales específicos en cribados de rebaños (6). Si bien esto es un reconocimiento, apuntar solo a los viromes no captura la naturaleza simbiótica de los virus y las bacterias que trabajan juntos para causar infección. Además, la NGS es oportuna y costosa, especialmente en un entorno de diagnóstico cuando el tiempo de respuesta de los resultados es crítico. Por lo tanto, el uso de herramientas como ONT MinION brinda la oportunidad de mitigar estas deficiencias y proporcionar una herramienta de diagnóstico rápido sin necesidad de conocimientos previos para la detección. Como un intento de desarrollar nuevas herramientas basadas en secuencias para la detección de patógenos, en este estudio, hemos desarrollado una plataforma que utiliza secuenciación de tercera generación (secuenciaciones de lectura larga utilizando la tecnología Oxford Nanopore) para identificar patógenos bacterianos y virales de especies ganaderas sin conocimientos previos.
4.1 La secuenciación de lectura larga permite la identificación de bacterias y virus de diferentes matrices sin conocimientos previos
Los métodos de diagnóstico basados en PCR y ELISA en tiempo real se utilizan ampliamente para el diagnóstico rápido y preciso de patógenos bacterianos y virales, o de respuestas inmunitarias a ellos. Sin embargo, para este tipo de tecnologías, se necesita información genética y molecular previa y no se detectan nuevos patógenos emergentes. Además, incluso en el caso de los patógenos con información genética conocida, los eventos de recombinación y mutación pueden hacer que los ensayos actuales no sean sensibles para permitir una detección precisa. Un buen ejemplo de ello es el brote de SARs-CoV-2 que demostró la aparición de mutaciones genéticas, lo que permitió la invasión de la detección (24). Sin embargo, el uso de enfoques de secuenciación del genoma completo ayuda a superar estos problemas. Kubacki et al. (6) demostraron el uso de la secuenciación de nueva generación (NGS) para la investigación de viromas en muestras clínicas de cerdos sin conocimiento previo, para identificar todos los virus que pueden estar presentes en una muestra determinada. Este estudio también informó sobre el uso de NGS para ayudar a proporcionar una comprensión más profunda de los virus, incluida la determinación de variantes genéticas. Sin embargo, Kubacki et al. (6) informaron que aunque la NGS proporciona muchos beneficios, como la falta de necesidad de información previa, precisión y análisis de variantes, el método requiere mucho tiempo y es costoso. Aquí hemos desarrollado una plataforma basada en NGS utilizando Oxford Nanopore Technologies para identificar patógenos virales y bacterianos, que podría llevarse a cabo en un período de tiempo de 12 horas desde la recolección de la muestra hasta el diagnóstico. El tiempo transcurrido desde la recogida de la muestra hasta el análisis de 12 h se calculó sobre la base de 2 h para la filtración, el tratamiento con DNasa y la extracción de ácidos nucleicos, 30 minutos para el control de calidad con Agilent BioAnalyzer 2000 (ARN y ADN), 4,5 h para la síntesis de ADNc ds, 3 h para la preparación de la biblioteca, 30 minutos para el control de calidad con Agilent BioAnalyzer y el ensayo de alta sensibilidad de fluorescencia Denovix solo para el ADN, y 1 h para la secuenciación. El análisis se puede realizar simultáneamente con la secuenciación en curso. El protocolo desarrollado se evaluó utilizando virus sustitutos introducidos en diferentes matrices de tejidos diagnósticamente relevantes.
El protocolo desarrollado es robusto y se puede utilizar con múltiples matrices de tejidos diferentes y utiliza tres enfoques diferentes para enriquecer los objetivos de patógenos virales y bacterianos. El enfoque desarrollado permite reducir la contaminación de la célula huésped utilizando filtros de 0,2 μm o 0,8 μm, lo que permite el enriquecimiento de partículas de virus (0,2 μm) o bacterias y partículas de virus (0,8 μm). Además, la concentración de muestras filtradas mediante ultracentrifugación aumenta la detección al concentrar las muestras. Por último, el tratamiento con ADNasa I y ARNasa A permite la eliminación del ADN y el ARN que flotan libremente de las células lisadas. Como tales, estos enfoques ayudan a reducir la contaminación del huésped y aumentan la sensibilidad, la reproducibilidad y la solidez de la detección de patógenos.
4.2 Validación y aplicabilidad del método para el diagnóstico y la vigilancia
Como informaron Kubacki et al. (6), los desafíos de los métodos basados en secuencias en comparación con los métodos convencionales basados en PCR en tiempo real para el diagnóstico incluyen el tiempo necesario para el procesamiento y análisis de muestras, lo que conduce a un retraso en el diagnóstico y la intervención. Aquí hemos reducido el tiempo de detección de 3 días, según lo reportado por Kubacki et al. (6), a 12 h. Además de los rápidos tiempos de respuesta, nuestro enfoque tiene una aplicación más amplia con la capacidad de detectar simultáneamente patógenos bacterianos y virales. Además, la canalización de análisis de datos personalizados impulsada por bioinformática permite analizar los datos en tiempo real.
Utilizando muestras desconocidas de animales con signos clínicos y muestras con detección confirmada de patógenos, intentamos validar la aplicabilidad del protocolo desarrollado para la identificación de patógenos. Con este fin, probamos el protocolo desarrollado contra virus de ADN y ARN y patógenos bacterianos de muestras de tejido porcino y bovino. Los patógenos se identificaron con éxito en estas muestras, lo que podría validar la aplicabilidad de este enfoque para su uso diagnóstico. Sin necesidad de información previa sobre el patógeno, este enfoque permite a los laboratorios disponer de una estrategia de identificación más sólida para identificar patógenos conocidos y desconocidos dentro de la misma muestra. A pesar de que no fue la intención dentro de este estudio, pudimos identificar otros reinos dentro de las muestras analizadas. Esta información podría proporcionar información valiosa para identificar rápidamente las especies indicadoras y los patógenos en las enfermedades polimicrobianas. Inesperadamente, observamos una alta abundancia de Mycoplasma en las muestras filtradas. Es probable que esta observación se debiera a que se descubrió que las líneas celulares utilizadas para propagar virus estaban contaminadas con Mycoplasma. No pudimos eliminar los micoplasmas debido al hecho de que son pequeños y carecen de una estructura de pared celular (25), y como resultado, postulamos, pudimos pasar a través del filtro utilizado para eliminar la contaminación celular del huésped. Sin embargo, incluso en presencia de contaminación por Mycoplasma, pudimos identificar suficientemente los virus sustitutos que se utilizaron para inocular los tejidos, lo que sugiere la solidez del protocolo desarrollado.
4.3 Profundidad de secuenciación y rentabilidad
Además de los métodos de procesamiento de muestras desarrollados, se desarrolló una canalización de análisis de datos en tiempo real para proporcionar resultados de análisis rápidos para la detección de patógenos. Para identificar la profundidad mínima de las lecturas de secuencia y la necesidad de una detección precisa para reducir el tiempo y el costo de la secuenciación, realizamos un análisis de los datos a medida que se liberan de la plataforma de secuenciación. Con este fin, analizamos conjuntos de datos generados en incrementos de 4.000 lecturas para las especies virales y bacterianas presentes dentro de la muestra de virus sustitutos que fueron inoculados. El análisis secuencial de las lecturas recuperadas a través de la secuenciación demostró claramente que una profundidad de lectura de aproximadamente 12.000 lecturas proporcionará una estimación sólida de los patógenos presentes en la muestra. Como tal, este enfoque reducirá el tiempo utilizado para la secuenciación (30 min) y los recursos utilizados para la secuenciación, reduciendo así los costos de diagnóstico. Además, en esta etapa, la ejecución se puede detener y la celda de flujo se puede reutilizar para una secuenciación adicional en un momento posterior. Como tal, el enfoque de código de barras descrito anteriormente aumenta el rendimiento de las muestras y ayuda a reducir el costo por muestra, lo que hace que el método desarrollado sea ampliamente aplicable a las muestras clínicas y a la vigilancia de patógenos actuales y emergentes.
La plataforma ONT MinION tiene ventajas como el rápido tiempo de respuesta, la portabilidad y la capacidad de detectar organismos en múltiples reinos. Además, la multiplexación ayuda a reducir el tiempo y el costo del análisis de muestras. Sin embargo, el costo del procesamiento de una sola muestra puede ser mayor que el de la PCR en tiempo real. Por lo tanto, si se utiliza para estudios de vigilancia, el costo puede ser prohibitivo.
Recientemente, la ONT ha desarrollado una herramienta, llamada ReadUntil, para la secuenciación selectiva, en la que si una lectura no coincide con los organismos objetivo de interés, la lectura puede ser expulsada por el poro. Aquí, las lecturas «no deseadas» se eliminarían del poro y una nueva lectura comenzaría a secuenciarse una vez que el poro se haya recuperado (9, 26). Según Kovaka et al. (9), esto se puede lograr invirtiendo la polaridad del voltaje a través del poro para expulsar la molécula de ADN y permitir que una nueva lectura de secuenciación comience antes. Además, si las lecturas no deseadas se identifican con la suficiente rapidez, el enriquecimiento de las lecturas de destino se puede realizar únicamente con una técnica computacional (9). En un estudio realizado por Ong et al. (27), se compararon la secuenciación ONT ReadUntil (adaptativa), la secuenciación ONT estándar y la secuenciación Illumina shotgun para identificar qué método era el más eficiente para perfilar el microbioma vaginal bovino y reducir las lecturas del huésped. Dentro de este estudio, se identificó un mayor número de genes anotados entre el grupo ONT ReadUntil (secuenciación adaptativa), lo que demostró las ventajas de la secuenciación adaptativa ONT en muestras que tienen una alta proporción de ADN huésped-microbio (27).
Además de ReadUntil, otro grupo ha optimizado el programa dentro de ONT y ha creado un programa adicional llamado UNCALLED que también optimiza aún más la reducción de la secuenciación de lecturas no deseadas de fragmentos más grandes mientras continúa refinándose durante la ejecución de la secuenciación (9). Dentro de este programa, los investigadores pueden cargar la secuenciación de las lecturas que desean enriquecer al permitir la continuación de la secuenciación dentro de un poro determinado o agotar la secuenciación de (lecturas no deseadas) por el poro que rechaza las lecturas no deseadas (9). Aquí, el algoritmo de Kovak et al. (9) convierte tramos de señales en k-mers y utiliza k-mers de mayor probabilidad como consulta para el índice de Ferragina-Manzini (FM) para buscar en bases de datos objetivo (9, 26).
Este enfoque puede ser útil para reducir los costos y centrarse en múltiples organismos objetivo de la misma muestra. Además, si la contaminación de la célula huésped es alta, el agotamiento de las lecturas del huésped podría permitir un mayor enriquecimiento de las lecturas microbianas. El uso de herramientas como ReadUntil o UNCALLED no permitirá la identificación de patógenos emergentes desconocidos. Por lo tanto, el enfoque descrito en nuestro estudio se puede aplicar a la detección de patógenos y organismos conocidos y desconocidos en un entorno determinado con la flexibilidad de modificar el protocolo de secuenciación en función de las necesidades del usuario.
5 Limitaciones
Si bien este protocolo es muy prometedor en su aplicabilidad para el diagnóstico clínico, el tipo de muestra y la calidad de los ácidos nucleicos extraídos podrían afectar el rendimiento. Como ONT MinION™ está desarrollado para la secuenciación de lectura larga, eso ayuda a mejorar las anotaciones, y la extracción de fragmentos cortos de ácido nucleico puede dificultar el rendimiento de este enfoque. Además, para muestras más complejas y muestras con mucha contaminación del huésped, el tratamiento con DNasa es fundamental y puede ser necesario realizarlo durante más tiempo o repetirlo. Para muestras más complejas y muestras con mayor contaminación del huésped, es necesario aumentar la profundidad de lectura. Una preocupación adicional es la calidad de lectura en la plataforma ONT MinION™. Sin embargo, los avances recientes en la tecnología han aumentado la precisión de las llamadas base del 98,3 % al ≥≥99% (28). Si bien ONT MinION™ permite la secuenciación en campo, el protocolo desarrollado en este estudio necesita un entorno de laboratorio y no es factible en el campo en su etapa actual.
6 Conclusión
Aquí desarrollamos un protocolo novedoso que utiliza la secuenciación de lectura larga para identificar patógenos bacterianos y virales conocidos y desconocidos del ganado que es rápido, robusto y puede aplicarse ampliamente. Además, hemos demostrado que el protocolo es robusto en el uso de patógenos conocidos y muestras clínicas con patógenos no caracterizados de diferentes tipos de tejidos y mostramos cómo se pueden utilizar las aplicaciones de esta tecnología para el diagnóstico y la vigilancia de enfermedades endémicas y emergentes del ganado.
Declaración de disponibilidad de datos
Las contribuciones originales presentadas en el estudio están a disposición del público. Estos datos se pueden encontrar en: https://www.ncbi.nlm.nih.gov/bioproject/; PRJNA1045613.
Declaración ética
El estudio en animales fue aprobado por el Comité de Uso y Cuidado de Animales de la Universidad de Nebraska. El estudio se llevó a cabo de acuerdo con la legislación local y los requisitos institucionales.
Contribuciones de los autores
AN: Curación de datos, Análisis formal, Investigación, Metodología, Redacción – borrador original, Redacción – revisión y edición. DL: Curación de datos, Análisis formal, Investigación, Metodología, Recursos, Redacción – revisión y edición. JD: Conceptualización, Curación de datos, Adquisición de fondos, Metodología, Administración de proyectos, Recursos, Redacción, revisión y edición. BB: Adquisición de fondos, Supervisión, Redacción, revisión y edición. SF: Conceptualización, Curación de datos, Obtención de fondos, Metodología, Administración de proyectos, Recursos, Supervisión, Redacción, revisión y edición.
Financiación
El/los autor/es declara(n) haber recibido apoyo financiero para la investigación, autoría y/o publicación de este artículo. Este proyecto fue financiado por la mejora de la Red Nacional de Laboratorios de Salud Animal de 2021, proyecto número AP20VDS y B000C008 y el Departamento de Agricultura de los Estados Unidos-Instituto Nacional de Alimentos y Agricultura, subvenciones n.º 2022-33522-38219 y 2023-68015-40015.
Reconocimientos
Los autores desean agradecer al Centro de Diagnóstico Veterinario de Nebraska por su contribución a las inoculaciones y extracciones de partículas virales.
Conflicto de intereses
SF, autor de esta publicación, ha revelado un interés financiero significativo en NuGut LLC. De acuerdo con su política de Conflicto de Intereses, el Comité de Conflicto de Intereses en Investigación de la Universidad de Nebraska Lincoln ha determinado que esto debe ser divulgado.
El resto de los autores declaran que la investigación se llevó a cabo en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un potencial conflicto de intereses.
El/los autor/es declararon, en el momento de la presentación, ser miembro del consejo editorial de Frontiers. Esto no tuvo ningún impacto en el proceso de revisión por pares ni en la decisión final.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o afirmación que pueda ser hecha por su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fvets.2024.1341783/full#supplementary-material
Notas
1. ^https://github.com/FernandoLab/MinION_high_consequence_viral_pathogens
Referencias
1. Andraud, M, y Rose, N. Modelización de enfermedades virales infecciosas en poblaciones porcinas: un estado del arte. Gestión de la Salud Porcina. (2020) 6:22. doi: 10.1186/s40813-020-00160-4
Resumen de PubMed | Texto completo de Crossref | Google Académico
2. Vander Waal, K. y J. Deen, tendencias globales en enfermedades infecciosas de cerdos. Proc Natl Acad Sci. (2018) 115:11495–500. doi: 10.1073/pnas.1806068115
Resumen de PubMed | Texto completo de Crossref | Google Académico
3. Organización de las Naciones Unidas para la Agricultura y la Alimentación (FA El estado de la inseguridad alimentaria en el mundo. Roma, Italia: FAO (2008).
4. Yu, W, y Jensen, JD. Implicaciones para la sostenibilidad del aumento de la demanda mundial de carne de cerdo. Frente de animación. (2022) 12:56–60. doi: 10.1093/af/vfac070
Resumen de PubMed | Texto completo de Crossref | Google Académico
5. McLean, RK y Graham, SP. El cerdo como huésped amplificador de virus zoonóticos nuevos y emergentes. Una sola salud. (2022) 14:100384. doi: 10.1016/j.onehlt.2022.100384
Resumen de PubMed | Texto completo de Crossref | Google Académico
6. Kubacki, J, Fraefel, C, y Bachofen, C. Implementación de la secuenciación de nueva generación para la identificación de virus en laboratorios de diagnóstico veterinario. J Vet Diagn Invest. (2021) 33:235–47. doi: 10.1177/1040638720982630
Resumen de PubMed | Texto completo de Crossref | Google Académico
7. Frymus, T, Belák, S, Egberink, H, Hofmann-Lehmann, R, Marsilio, F, Addie, DD, et al. Infecciones por el virus de la influenza en gatos. Virus. (2021) 13:1435. doi: 10.3390/v13081435
8. Petersen, LM, Martin, IW, Moschetti, WE, Kershaw, CM, y Tsongalis, GJ. Secuenciación de tercera generación en el laboratorio clínico: explorando las ventajas y los retos de la secuenciación de nanoporos. J Clin Microbiol. (2019) 58:19. doi: 10.1128/JCM.01315-19
Resumen de PubMed | Texto completo de Crossref | Google Académico
9. Kovaka, S, Fan, Y, Ni, B, Timp, W y Schatz, MC. Secuenciación dirigida de nanoporos mediante el mapeo en tiempo real de la señal eléctrica sin procesar con UNCALLED. Nat Biotechnol. (2021) 39:431–41. doi: 10.1038/s41587-020-0731-9
Resumen de PubMed | Texto completo de Crossref | Google Académico
10. Bracht, AJ, O’Hearn, ES, Fabian, AW, Barrette, RW y Sayed, A. Ensayo de PCR con transcripción inversa en tiempo real para la detección de Senecavirus a en muestras de diagnóstico vesicular porcina. PLoS Uno. (2016) 11:e0146211. doi: 10.1371/journal.pone.0146211
Resumen de PubMed | Texto completo de Crossref | Google Académico
11. Fowler, VL, Ransburgh, RH, Poulsen, EG, Wadsworth, J, King, DP, Mioulet, V, et al. Desarrollo de un novedoso ensayo de RT-PCR en tiempo real para detectar el virus Seneca Valley-1 asociado a casos emergentes de enfermedad vesicular en cerdos. Métodos J Virol. (2017) 239:34–7. doi: 10.1016/j.jviromet.2016.10.012
Resumen de PubMed | Texto completo de Crossref | Google Académico
12. Horwood, PF, y Mahony, TJ. Detección múltiple en tiempo real de RT-PCR de tres virus asociados al complejo de enfermedades respiratorias bovinas. Métodos J Virol. (2011) 171:360–3. doi: 10.1016/j.jviromet.2010.11.020
Resumen de PubMed | Texto completo de Crossref | Google Académico
13. Liang, H, Geng, J, Bai, S, Aimuguri, A, Gong, Z, Feng, R, et al. PCR en tiempo real para la detección del virus de la diarrea viral bovina. Pol J Vet Sci. (2019) 22:405–13. doi: 10.24425/pjvs.2019.129300
Resumen de PubMed | Texto completo de Crossref | Google Académico
14. Mahlum, CE, Haugerud, S, Shivers, JL, Rossow, KD, Goyal, SM, Collins, JE, et al. Detección del virus de la diarrea viral bovina mediante la reacción en cadena de la polimerasa con transcriptasa inversa de Taq man. J Vet Diagn Invest. (2002) 14:120–5. doi: 10.1177/104063870201400205
15. Richt, JA, Lager, KM, Clouser, DF, Spackman, E, Suárez, DL y Yoon, KJ. Ensayos de reacción en cadena de la polimerasa con transcriptasa inversa en tiempo real para la detección y diferenciación de los virus de la influenza porcina de América del Norte. J Vet Diagn Invest. (2004) 16:367–73. doi: 10.1177/104063870401600501
Resumen de PubMed | Texto completo de Crossref | Google Académico
16. Thonur, L, Maley, M, Gilray, J, Crook, T, Laming, E, Turnbull, D, et al. RT-PCR multiplexada en tiempo real de un solo paso para la detección del virus respiratorio sincitial bovino, el herpesvirus bovino 1 y el virus de la parainfluenza bovina 3. BMC Vet Res. (2012) 8:37. doi: 10.1186/1746-6148-8-37
Resumen de PubMed | Texto completo de Crossref | Google Académico
17. Wang, J, O’Keefe, J, Orr, D, Loth, L, Banks, M, Wakeley, P, et al. Validación de un ensayo de PCR en tiempo real para la detección de herpesvirus bovino 1 en semen bovino. Métodos J Virol. (2007) 144:103–8. doi: 10.1016/j.jviromet.2007.04.002
Resumen de PubMed | Texto completo de Crossref | Google Académico
18. Stangegaard, M, Dufva, IH y Dufva, M. La transcripción inversa utilizando cebadores pentadecámeros aleatorios aumenta el rendimiento y la calidad del ADNc resultante. BioTécnicas. (2006) 40:649–57. doi: 10.2144/000112153
Resumen de PubMed | Texto completo de Crossref | Google Académico
19. Wick, RR, Judd, LM, Gorrie, CL y Holt, KE. Completar los ensamblajes del genoma bacteriano con secuenciación multiplex min ION. Genoma Microb. (2017) 3:E000132. doi: 10.1099/mgen.0.000132
Resumen de PubMed | Texto completo de Crossref | Google Académico
20. Kim, D, Song, L, Breitwieser, FP, y Salzberg, SL. Centrífuga: clasificación rápida y sensible de secuencias metagenómicas. Genoma Res. (2016) 26:1721–9. doi: 10.1101/gr.210641.116
Resumen de PubMed | Texto completo de Crossref | Google Académico
21. Breitwieser, FP, y Salzberg, SL. Pavian: análisis interactivo de datos metagenómicos para estudios de microbioma e identificación de patógenos. Bioinformática. (2020) 36:1303–4. doi: 10.1093/bioinformatics/btz715
Resumen de PubMed | Texto completo de Crossref | Google Académico
22. Brunborg, IM, Moldal, T y Jonassen, CM. Cuantificación del circovirus porcino tipo 2 aislado de muestras de suero/plasma y tejido de cerdos sanos y cerdos con síndrome de desgaste multisistémico postdestete mediante una PCR en tiempo real basada en Taq man. Métodos J Virol. (2004) 122:171–8. doi: 10.1016/j.jviromet.2004.08.014
23. Zsak, L, Borca, MV, Risatti, GR, Zsak, A, French, RA, Lu, Z, et al. Diagnóstico preclínico de la peste porcina africana en cerdos expuestos al contacto mediante un ensayo de PCR en tiempo real. J Clin Microbiol. (2005) 43:112–9. doi: 10.1128/JCM.43.1.112-119.2005
Resumen de PubMed | Texto completo de Crossref | Google Académico
24. Zelyas, N, Pabbaraju, K, Croxen, MA, Lynch, T, Buss, E, Murphy, SA, et al. Respuesta de precisión al aumento de la variante preocupante B.1.1.7 del SARS-CoV-2 mediante la combinación de nuevos ensayos de PCR y secuenciación del genoma para la detección y vigilancia rápidas de variantes. Microbiología. Espectro. (2021) 9:E00315–21. doi: 10.1128/Spectrum.00315-21
25. Pereyre, S, Goret, J y Bébéar, C. Mycoplasma pneumoniae: conocimiento actual sobre la resistencia y el tratamiento de los macrólidos. Microbiol frontal. (2016) 7:974. doi: 10.3389/fmicb.2016.00974
26. Martin, S, Heavens, D, Lan, Y, Horsfield, S, Clark, MD y Leggett, RM. Muestreo adaptativo de nanoporos: una herramienta para el enriquecimiento de especies de baja abundancia en muestras metagenómicas. Genoma Biol. (2022) 23:11. doi: 10.1186/s13059-021-02582-x
Resumen de PubMed | Texto completo de Crossref | Google Académico
27. Ong, CT, Ross, EM, Boe-Hansen, GB, Turni, C, Hayes, BJ, y Tabor, AE. Nota técnica: superación de la contaminación del huésped en muestras metagenómicas vaginales bovinas con secuenciación adaptativa de nanoporos. J Anim Sci. (2021) 100:344. doi: 10.1093/jas/skab344
Palabras clave: brotes, tecnología Oxford Nanopore MinION™, virus, vigilancia, porcino
Cita: Neujahr AC, Loy DS, Loy JD, Brodersen BW y Fernando SC (2024) Detección rápida de patógenos virales emergentes y de alta consecuencia en cerdos. Frente. Vet. Sci. 11:1341783. doi: 10.3389/fvets.2024.1341783
Editado por:
Marta Canuti, Universidad de Copenhague, Dinamarca
Revisado por:
Lia Van Der Hoek, Universidad de Ámsterdam, Países Bajos
Jenni Virtanen, Universidad de Helsinki, Finlandia
Derechos de autor © 2024 Neujahr, Loy, Loy, Brodersen y Fernando. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia Creative Commons Attribution License (CC BY).
*Correspondencia: Samodha C. Fernando, sfernando2@unl.edu
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o afirmación que pueda hacer su fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía