
Evaluación de la estabilidad del genoma de la cepa de la vacuna contra el PRRS con un nuevo protocolo de secuenciación de estilo ARTIC

Evaluación de la estabilidad del genoma de la cepa de la vacuna contra el PRRS con un nuevo protocolo de secuenciación de estilo ARTIC
 Szilvia Jakab1,2
Szilvia Jakab1,2  Ádám Bálint3
Ádám Bálint3  Karolina Cseri4,5 Krisztina Bali1,2
Karolina Cseri4,5 Krisztina Bali1,2  Eszter Kaszab1,2,4
Eszter Kaszab1,2,4  Marianna Domán1,2 Máté Halas6 Krisztina Szarka4,5
Marianna Domán1,2 Máté Halas6 Krisztina Szarka4,5  Krisztián Bányai1,2,7*
Krisztián Bányai1,2,7*- 1Grupo de Descubrimiento de Patógenos, Instituto de Investigación Médica Veterinaria HUN-REN, Budapest, Hungría
- 2Laboratorio Nacional de Enfermedades Infecciosas de los Animales, Resistencia a los Antimicrobianos, Salud Pública Veterinaria y Seguridad de la Cadena Alimentaria, Budapest, Hungría
- 3Dirección de Diagnóstico Veterinario, Oficina Nacional de Seguridad de la Cadena Alimentaria, Budapest, Hungría
- 4One Health Institute, Universidad de Debrecen, Debrecen, Hungría
- 5Departamento de Metagenómica, Universidad de Debrecen, Debrecen, Hungría
- 6Prophyl Ltd., Mohács, Hungría
- 7Departamento de Farmacología y Toxicología, Universidad de Medicina Veterinaria, Budapest, Hungría
Se desarrolló un protocolo de secuenciación de amplicones en mosaico para analizar la estabilidad de la secuencia genómica de la cepa viva modificada de la vacuna contra el PRRSV, Porcilis MLV. La columna vertebral del protocolo de estilo ARTIC estaba formada por 34 pares de cebadores individuales, que se dividieron en dos grupos de cebadores. Los pares de cebadores se diseñaron para amplificar fragmentos de 532 a 588 pb de la región genómica correspondiente. Los amplicones son adecuados para la secuenciación en secuenciadores de ADN Illumina con kits de secuenciación de 600 ciclos disponibles. La concentración de pares de cebadores en los grupos se optimizó para obtener una profundidad de secuenciación equilibrada a lo largo del genoma. También se analizaron los datos de secuenciación profunda de tres lotes de vacunas. Los tres lotes de vacunas eran muy similares entre sí, aunque también mostraban variaciones de un solo nucleótido (SNV) que afectaban a menos del 1 % del genoma. En las tres cepas vacunales, se identificaron entre 113 y 122 sitios SNV; En estos sitios, las variantes minoritarias representaron un rango de frecuencia del 1 al 48.7 por ciento. Además, las cepas dentro de los lotes contenían polimorfismos de longitud bien conocidos; Los genomas de estos mutantes minoritarios de deleción eran de 135 a 222 pb más cortos que los de la variante con el genoma completo. Nuestros resultados muestran la utilidad de los protocolos de estilo ARTIC en la evaluación de la estabilidad genómica de cepas de PRRS MLV.
1 Introducción
El síndrome reproductivo y respiratorio porcino (PRRS) sigue siendo una de las enfermedades víricas más devastadoras en los sistemas de producción porcina, responsable de graves pérdidas económicas en todo el mundo. Las especies virales causantes, Betaarterivirus suid 1 y Betaarterivirus suid 2 (también conocidas como PRRSV-1 y -2, respectivamente) son virus de ARN monocatenario envueltos, de sentido positivo, con un genoma de aproximadamente 15 mil nucleótidos (nt) de longitud (1-3). El PRRSV se caracteriza por una alta diversidad genética, lo que plantea desafíos para las medidas de control de la enfermedad (4, 5).
Las vacunas de virus vivos modificados (MLV, por sus siglas en inglés) son herramientas ampliamente utilizadas para la prevención y el control del PRRS. Estas vacunas se han desarrollado para reducir la gravedad clínica y la diseminación del virus, aliviando la carga económica asociada a la enfermedad. Desde su primera introducción en el año 2000, las vacunas contra el PRRS (MSD Animal Health) contra el PRRSV-1 han sido las vacunas preferidas para la inmunización activa de cerdas y cerdos en crecimiento (6). En general, el monitoreo de la seguridad de las vacunas es importante durante la fabricación, sin embargo, la información sobre la estabilidad genética de las cepas de vacunas en diferentes lotes es limitada (7, 8). La naturaleza cuasiespecie de las vacunas basadas en virus vivos plantea desafíos adicionales. Hasta la fecha, se ha determinado la secuencia completa del genoma del virus de cuatro lotes diferentes de Porcilis PRRS MLV, lo que muestra que algunas variantes de deleción coexisten en diferentes lotes de vacunas (9). Además de la estabilidad de la vacuna, otra cuestión a considerar es la pérdida de atenuación de las cepas de MLV en el campo que ya se ha confirmado para algunos MLV del PRRS de uso común (9-14). Independientemente de la posibilidad teórica de reversión de la cepa vacunal, los datos indican que los aislados de campo derivados de Porcilis son genéticamente más estables, al menos según el análisis de secuencias ORF5 y ORF7, que algunas otras vacunas (15).®®
La estabilidad genética de las vacunas vivas es un requisito fundamental para su desarrollo, producción y uso en el campo. La secuenciación del genoma completo en plataformas de secuenciación de nueva generación permite una descripción más precisa de la estructura poblacional de las cepas vacunales. La secuenciación en mosaico de los protocolos de secuenciación de amplicones podría superar los problemas de sensibilidad, ya que la secuenciación de nueva generación está precedida por la amplificación dirigida del genoma utilizando un gran conjunto de pares de cebadores. Este enfoque se ha vuelto muy popular desde el comienzo de la pandemia de SARS-CoV-2. La red ARTIC ha desarrollado numerosos protocolos para la amplificación directa de genomas de virus a partir de muestras clínicas utilizando cebadores multiplexados en mosaico1 (16). En 2020, Gohl y sus colaboradores desarrollaron aún más el enfoque y establecieron un método basado en amplicones de cola para la preparación de bibliotecas de ADN y la secuenciación de NGS (17). Dado que los cebadores específicos de la diana se pueden personalizar fácilmente, este método puede adaptarse teóricamente a cualquier genoma viral, ofreciendo un enfoque rápido, sensible y económico para la secuenciación del genoma completo.
Los objetivos de este estudio fueron el desarrollo de un método de secuenciación de amplicones de cola y mosaico específico para la cepa Porcilis PRRS MLV y la exploración de la estabilidad genética de lotes comerciales de vacunas Porcilis PRRS. Este método de secuenciación simple, rápido y rentable podría servir como una herramienta fácilmente adaptable para afirmar la calidad de los MLV del PRRS.®®
2 Materiales y métodos
2.1 Vacunas
Se utilizaron tres lotes diferentes de la vacuna Porcilis PRRS (lote n.º A220CE01, A220AD01 y A221AE01) para desarrollar un protocolo ARTIC y comparar la estabilidad genética de la cepa Porcilis MLV.®
2.2 Extracción de ARN y síntesis de ADNc
Los lotes de vacunas se diluyeron a 1:8 en PBS y luego el ARN viral se aisló con el kit de virus de ARN NucleoSpin (Macherey-Nagel, Düren, Alemania) de acuerdo con las instrucciones del fabricante.
La transcripción inversa cebada aleatoriamente se realizó utilizando el SuperScript™ IV VILO™ Master Mix (Thermo Fisher Scientific, Waltham, MA, Estados Unidos) de acuerdo con las instrucciones del fabricante. La mezcla de reacción RT consistió en 4 μL de mezcla maestra (con oligo (dT) 18 y cebadores de hexámero aleatorios incluidos), 11 μL de agua libre de nucleasa y se añadió 5 μL de molde de ARN. El perfil térmico de la reacción fue el siguiente: la etapa de desnaturalización se realizó a 65 °C durante 5 min, luego el recocido de los cebadores, la transcripción inversa y la inactivación de la enzima duraron 10 min a 25 °C, 10 min a 50 °C y 5 min a 85 °C, respectivamente.
2.3 Diseño de la imprimación
Se diseñó un esquema de amplicones de mosaico para la amplificación del genoma completo. Nuestro objetivo era crear un conjunto de cebadores robusto específico para la cepa de la vacuna Porcilis y adecuado para su uso en PCR múltiples. Por lo tanto, recopilamos registros del genoma completo del GenBank que mostraron una identidad de secuencia de nucleótidos del >95% a la cepa de referencia de Porcilis DV (n.º de accesión, KJ127878). Este umbral se eligió en base a la observación de que las cepas derivadas de Porcilis muestran aproximadamente este rango de diversidad cuando se aíslan en condiciones de campo; En este sentido, esperábamos que las condiciones de producción de vacunas no permitieran un alto grado de diversificación. Las secuencias del genoma completo se alinearon mediante el algoritmo MAFFT en el software Geneious Prime (versión 2022.2.2) y se extrajo la secuencia de consenso (umbral: 90%) y se utilizó para el diseño del cebador. El conjunto de cebadores se diseñó en el servidor web PrimalScheme dando la secuencia de consenso como entrada (16).®
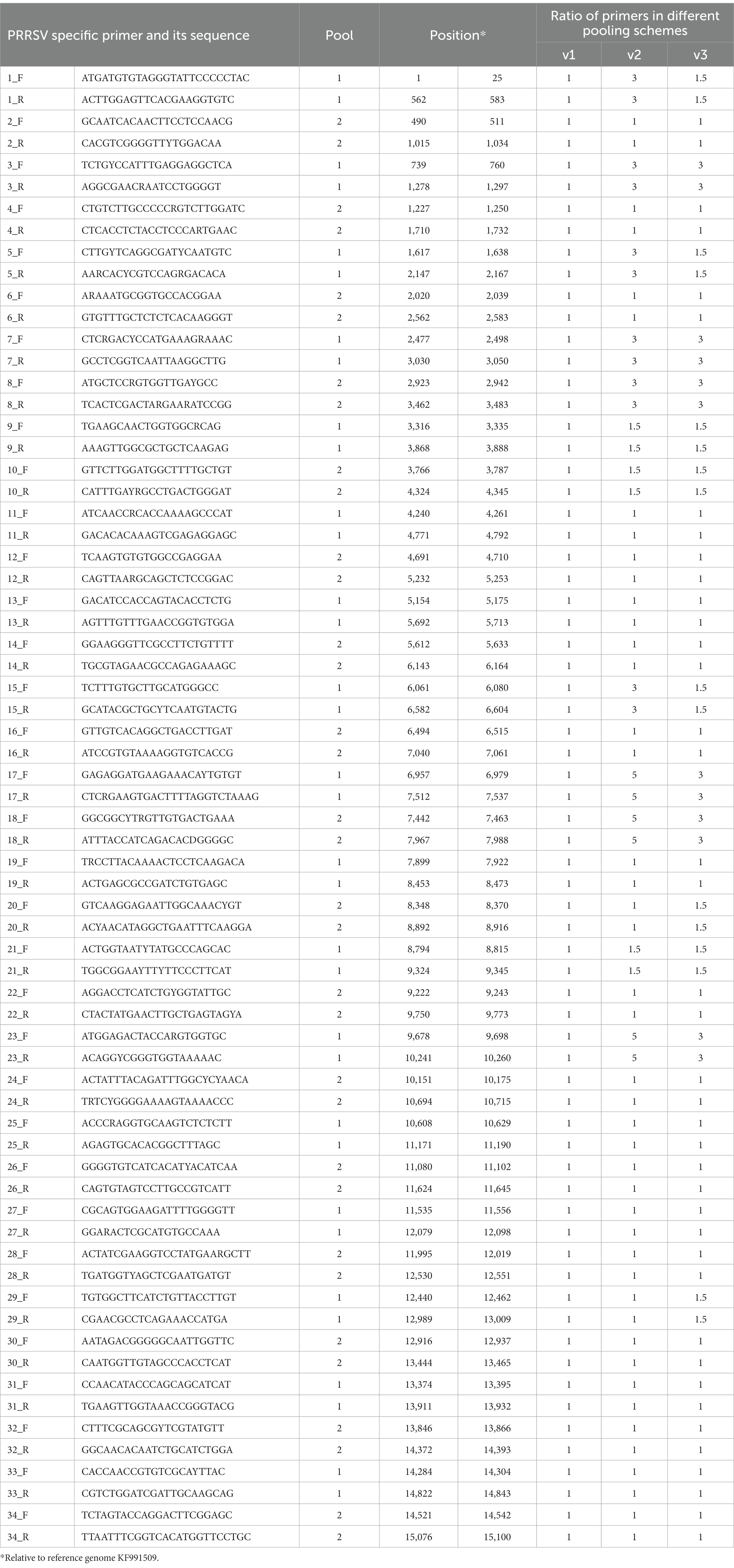 Tabla 1. Características de los cebadores de PRRSV probados en este estudio.
Tabla 1. Características de los cebadores de PRRSV probados en este estudio.
adelante: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG <cartilla específica del PRRSV>
y a la inversa: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG <imprimación específica del PRRSV>. Los cebadores de indexación directa e inversa compatibles con Illumina contenían parte de los sitios de unión del cebador de secuenciación, los índices i5 e i7 (Nextera XT Index Kit v2 Set C) y los adaptadores P5 y P7, respectivamente.
(F: 5′-AATGATACGGCGACCGAGATCTACACi5TCGTCGGCAGCGTC-3‘,
R: 5′-CAAGCAGAAGACGGCATACGAGATi7GTCTCGTGGGCTCGG-3‘).
Para generar los grupos de cebadores, los cebadores se resuspendieron a una concentración de stock de 100 μM y luego se dividieron en dos grupos (poolAv1, del grupo A1 y A2, respectivamente) combinando el mismo volumen de amplicones pares e impares. Se prepararon otros dos esquemas de agrupación, poolAv2 y poolAv3, para ajustar las proporciones relativas de cebadores y, en consecuencia, amplicones, para lograr una cobertura más equilibrada del genoma.
2.4 Preparación de la biblioteca y secuenciación de nueva generación
La amplificación del genoma completo relacionada con la preparación de la biblioteca consistió en un protocolo de PCR de dos pasos. Ambas rondas de PCR (PCR-1 y PCR-2) se realizaron utilizando la ADN polimerasa de alta fidelidad Q5U Hot Start (NEB, Frankfurt, Alemania) de acuerdo con las instrucciones del fabricante. Las reacciones de PCR-1 separadas para el conjunto de cebadores multiplex de los grupos A1 y A2 se compusieron de 15,46 μL de agua libre de nucleasa, 5 μL de tampón de reacción Q5U 5X, 0,5 μL de mezcla dNTP (10 mM), 1,29 μL de cebador A1 o A2 (10 μM, concentración final de 0,015 μM por cebador), 0,25 μL de ADN polimerasa de alta fidelidad Q5U Hot Start y 2,5 μL de plantilla de ADNc. Los ciclos de PCR se realizaron a 98 °C durante 30 s, seguidos de 30 ciclos de 98 °C durante 15 s y 65 °C durante 5 min, luego se mantuvieron a 4 °C. Los productos de PCR-1 se analizaron en gel de agarosa al 1%, las bandas se extirparon a 600 pb más o menos, y el ADN se purificó a partir de rodajas de gel con el kit de extracción de fragmentos de ADN de Gel/PCR (Favorgen, Ping Tung, Taiwán). Después de la extracción en gel, los grupos A1 y A2 se combinaron y luego se diluyeron 1:100 para cada muestra. Durante la segunda PCR, se agregaron adaptadores e índices específicos de Illumina a los productos PCR-1. PCR-2 compuesta por 11,75 μL de agua libre de nucleasa, 5 μL de tampón de reacción 5X Q5U, 0,5 μL de mezcla dNTP (10 mM), 1,25-1,25 μL de cebadores de indexación directa e inversa (10 μM), 0,25 μL de ADN polimerasa de alta fidelidad Q5U Hot Start y 5 μL de plantilla de ADN agrupada y diluida de PCR-1. Los ciclos de PCR se realizaron a 98 °C durante 30 s, 30 ciclos de 98 °C durante 20 s, 55 °C durante 15 s y 72 °C durante 1 min, la extensión final duró 5 min a 72 °C, luego se mantuvo a 4 °C. Una vez más, los productos de PCR-2 se analizaron en gel de agarosa al 1%, las bandas se extirparon a ~600 pb y las rodajas de gel se aislaron con el kit de extracción de fragmentos de ADN en gel/PCR (Favorgen, Ping Tung, Taiwán).®
Las concentraciones de las bibliotecas de ADN se midieron utilizando un equipo Qubit y luego se diluyeron a 4 nM. Este grupo se desnaturalizó y luego se diluyó a 20 pM, se enriqueció con un 35% de PhiX y se secuenció en un secuenciador Illumina Miseq utilizando el kit de reactivos v3 (600 ciclos) (Illumina, San Diego, CA, Estados Unidos).
2.5 Análisis de datos de secuencia
En primer lugar, las lecturas de secuencia de Illumina se recortaron de la siguiente manera: se descartaron las lecturas de secuencia inferiores a 75 pb, las secuencias de cebadores específicas de PRRSV y las bases de baja calidad (puntuación mínima de Phred de 10) se eliminaron en ambos extremos utilizando el complemento BBDuk en Geneious Prime. En segundo lugar, el ensamblaje del genoma se realizó en Geneious Prime con el algoritmo Geneious incorporado, o si fue necesario, con el ensamblador Minimap2, implementando el mapeo de referencia a una cepa de Porcilis DV (n.º de acceso, KF991509).®®
La variabilidad de la secuencia intracepa se analizó a partir de los datos de lectura corta de poolAv3. Para el análisis de las variantes de un solo nucleótido (SNVs) se descartaron las lecturas de secuencia inferiores a 75 pb, se eliminaron las secuencias de cebadores específicos del PRRSV y las bases de baja calidad (puntuación mínima de Phred de 30) en ambos extremos utilizando el plugin BBDuk en Geneious Prime. Utilizamos el algoritmo incorporado de Geneious Prime para la llamada de variantes con una frecuencia mínima de variantes del 1%. Solo se aceptaron los SNV que se identificaron en secuencias paralelas.®®
3 Resultados y discusión
3.1 Evaluación del ensayo
En este estudio, desarrollamos y probamos un protocolo de secuenciación de amplicones de mosaico específico para una cepa de vacuna contra el PRRSV ampliamente utilizada, Porcilis MLV. El flujo de trabajo se basó en un método de preparación de la biblioteca de PCR de dos pasos. En primer lugar, examinamos si el conjunto de cebadores diseñado amplifica con éxito diferentes lotes de Porcilis MLV. En este sentido, nos encontramos con un obstáculo inmediato, ya que el estudio se puso en marcha después de que Hungría fuera declarada libre de PRRS y la vacuna Porcilis MLV estuviera disponible en cantidades limitadas en el mercado. Además de la evaluación de las combinaciones de cebadores individuales, nos centramos en la mejora del equilibrio de cebadores en grupos de cebadores individuales para lograr una mejor cobertura de secuencias en todo el genoma.
En el primer paso del desarrollo de la prueba, nuestros resultados mostraron que los 34 pares de cebadores generaron productos de PCR del tamaño esperado en las reacciones separadas (Figura 1). A continuación, estos productos de PCR se agruparon para obtener relaciones equimolares para cada amplicón (1 a 34) y luego se sometieron a secuenciación de estos grupos. La profundidad total de secuenciación osciló entre 9.251x y 13.889x y el contig ensamblado cubrió el genoma completo (Figura 2). Estos datos indicaron que el diseño del ensayo cumplió con nuestras expectativas. Sin embargo, debido a que no era práctico utilizar los pares de cebadores en tubos de reacción separados, los cebadores se agruparon. En esta práctica, seguimos los protocolos previamente establecidos para otros virus y combinamos los pares pares pares e impares de cebadores en tubos de reacción separados (poolAv1) (16, 17). Por lo tanto, se prepararon dos mezclas de cebadores y las plantillas de ADNc se amplificaron en las mismas condiciones en estos dos tubos de reacción durante la ronda de PCR-1.
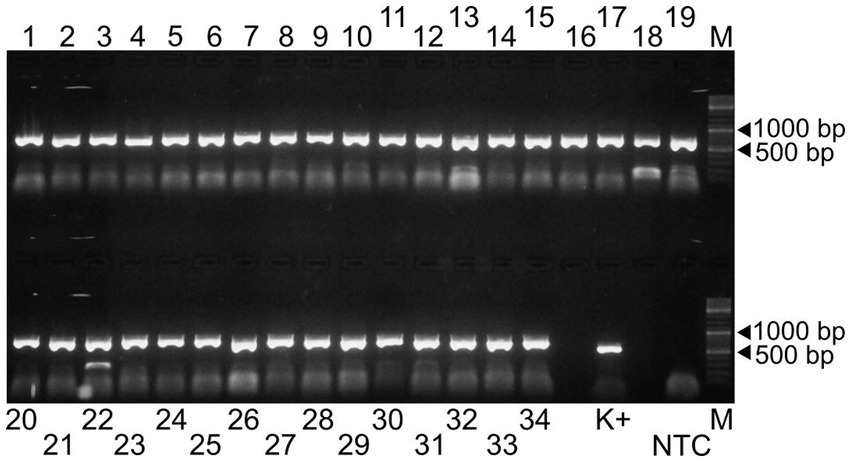 Figura 1. Electroforesis en gel de agarosa de los 34 amplicones generados por los pares de cebadores específicos de Porcilis MLV (carriles 1 a 34). El producto de PCR obtenido por un par de cebadores de diagnóstico dirigidos a la región ORF7 se utilizó como control positivo (K+) (18). NTC y M se refieren al control sin plantilla y a la mezcla de escalera de ADN de GeneRuler, respectivamente.
Figura 1. Electroforesis en gel de agarosa de los 34 amplicones generados por los pares de cebadores específicos de Porcilis MLV (carriles 1 a 34). El producto de PCR obtenido por un par de cebadores de diagnóstico dirigidos a la región ORF7 se utilizó como control positivo (K+) (18). NTC y M se refieren al control sin plantilla y a la mezcla de escalera de ADN de GeneRuler, respectivamente.
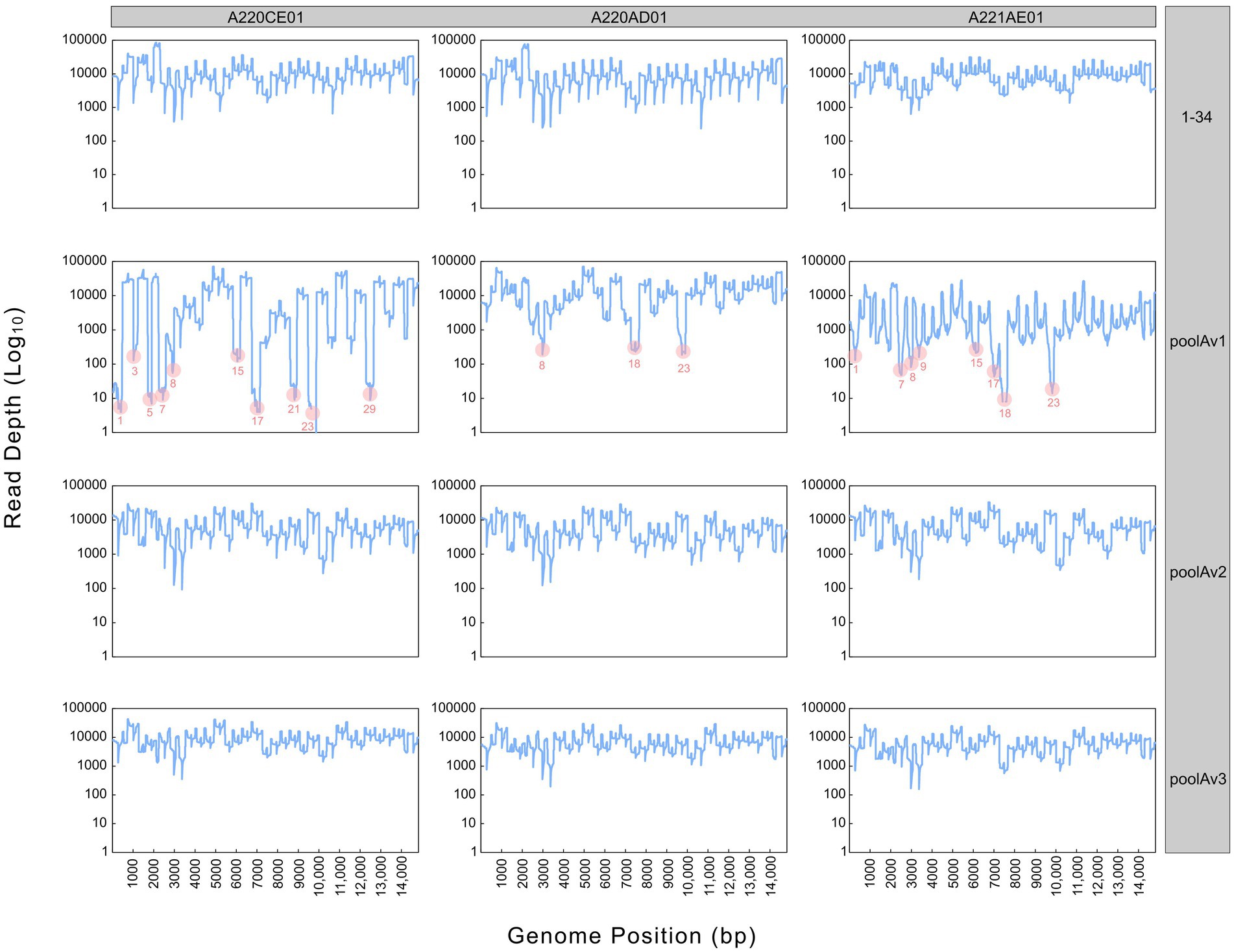 Figura 2. Profundidad de secuenciación derivada de diferentes enfoques de agrupación (1–34, poolAv1, poolAv2, poolAv3) determinada para los tres lotes de vacunas (A220CE01, A220AD01, A221AE01). En las parcelas vinculadas a poolAv1, se destacan los amplicones de caída.
Figura 2. Profundidad de secuenciación derivada de diferentes enfoques de agrupación (1–34, poolAv1, poolAv2, poolAv3) determinada para los tres lotes de vacunas (A220CE01, A220AD01, A221AE01). En las parcelas vinculadas a poolAv1, se destacan los amplicones de caída.
La secuenciación de la cepa de la vacuna Porcilis de diferentes lotes fue exitosa con poolAv1 (Figura 2). La cobertura del genoma, dependiendo del lote de vacunas, se acercó o alcanzó el 100%, mientras que la profundidad de secuenciación varió dentro y entre todos los lotes de vacunas. Observamos una reducción de la profundidad de secuenciación en los amplicones 1, 3, 5, 7, 8, 15, 17, 21, 23 y 29 para A220CE01, en los amplicones 8, 18 y 23 para A220AD01, y en los amplicones 1, 7, 8, 9, 15, 17, 18 y 23 para A221AE01, respectivamente (Figura 2). Por lo tanto, 7 de los 12 pares de cebadores dieron como resultado una reducción de la cantidad de sus respectivos productos de PCR en al menos dos lotes de vacunas distintos. Además, en el caso del amplicón 23, no se generaron lecturas en A220CE01. Para evaluar mejor la eficiencia de los pares de cebadores, calculamos la profundidad media de secuenciación por amplicón y por muestra de vacuna excluyendo las regiones de amplicones superpuestas. La profundidad media de secuenciación fue de 9.944x, 11.069x y 1799x, mientras que el rango de profundidades de secuenciación osciló entre 3x y 39.624x, 290x y 37.921x, y 12x y 18.107x para A220CE01, A220AD01 y A221AE01, respectivamente. Las diferentes eficiencias de los pares de cebadores utilizados en la relación equimolar nos impulsaron a optimizar el ensayo.
Para mejorar el equilibrio de los cebadores, modificamos la concentración de trabajo de los cebadores seleccionados en las mezclas de reacción y preparamos dos concentraciones de cebadores experimentales distintas (Tabla 1). Como tal, preparamos otras dos variaciones de poolA, designadas como v2 y v3. La Figura 2 muestra la cobertura y la profundidad de secuenciación logradas mediante el uso de grupos de cebadores con diferentes proporciones de concentración de cebadores individuales. Al usar poolAv2 y poolAv3, alcanzamos una profundidad de secuenciación más equilibrada a lo largo de todo el genoma, en comparación con poolAv1. Por ejemplo, las diferencias entre la profundidad mínima y máxima por amplicón alcanzada por poolAv3 para los tres lotes de vacunas fue de 14 a 16 veces en comparación con la diferencia de 131 a 14.151 veces observada cuando se usaron cebadores en proporciones equimolares (poolAv1). En general, la optimización de la concentración del cebador dio como resultado una disminución significativa del número de regiones de baja profundidad y, al mismo tiempo, una disminución de las regiones sin lecturas de secuencia. Como resultado, logramos una profundidad de secuenciación más equilibrada para todo el genoma. La necesidad de optimizar las concentraciones de cebadores ya se demostró para otros protocolos de secuenciación del genoma completo basados en amplicones de mosaico, por ejemplo, para los cebadores ARTIC para SARS-CoV-2 ampliamente utilizados (19).
3.2 Variabilidad de la cepa vacunal
Al comparar las secuencias de consenso generadas por los cuatro enfoques de agrupación diferentes, como la PCR-1 separada con 34 pares de cebadores y la agrupación A con los tres esquemas de agrupación diferentes, identificamos algunas diferencias de nucleótidos en las secuencias genómicas de consenso ensambladas. En el genoma de consenso de A220CE01 de poolAv1 y 1-34, en A220AD01 de poolAv2 y en A221AE01 de poolAv1 identificamos, respectivamente, diferencias de 6 nt y 2 nt, 1 nt y 5 nt con respecto a los demás lotes dados. Entre estas, 1 (A220CE01), 1 (A220AD01) y 3 (A221AE01) mutaciones correspondieron a los sitios identificados del SNV. Para explorar más a fondo la variación de la cepa de la vacuna, se utilizaron secuencias de lectura corta generadas por el poolAv3 para los análisis genómicos.
Anteriormente, se encontraron tres variantes diferentes de deleción de la cepa Porcilis en algunos lotes de vacunas; estos se designaron como LONG-DEL, SHORT-DEL y SHIFT-DEL. La diferencia entre las tres variantes se observó en las regiones genómicas 2.216 a 2.437 para LONG-DEL, 2.231 a 2.365 para SHORT-DEL, 2.344 a 2.435 y 2.446 a 2.506 para SHIFT-DEL (9). Al analizar los datos de poolAv3, además de la variante FULL-LENGTH, también pudimos detectar todas las variantes de deleción en los tres lotes de vacunas, y estimamos que la variante LONG-DEL era la forma más abundante de la cepa de la vacuna Porcilis, un hallazgo que se corresponde con los resultados publicados anteriormente (9). Los análisis genéticos posteriores se realizaron con la variante LONG-DEL. En resumen, las tres variantes genómicas LONG-DEL ensambladas de la cepa de la vacuna Porcilis tenían una longitud de 14.853 nt.
Los sitios 1_F (5′-ATGATGTGTAGGGTATTCCCCCCCCCCCCCTAC-3′) y 34_R (5′-TTAATTTCGGTCACATGGTTCCTGC-3′) en los extremos 5′ y 3′ del genoma, respectivamente, se excluyeron del ensamblaje, ya que no se generaron amplicones superpuestos en esta región.
Los genomas completos derivados de los tres lotes de vacunas fueron casi idénticos, solo se detectó un sitio polimorfo en la posición 1.519 (la posición es relativa a la cepa Porcilis DV; n.º de acceso KF991509). La identidad de nucleótidos por pares de todos los genomas completos de Porcilis disponibles de diferentes lotes de vacunas, incluidas las cepas de este estudio, varió entre el 99,9 y el 100% (la región delecionada en la posición nt 2.215-2.506 se excluyó de la alineación y los cálculos). Cuando se compararon las cepas vacunales con la cepa DV de tipo salvaje (n.º de accesión, KJ127878), los valores de identidad nucleotídica alcanzaron el 99,1%. De hecho, observamos un total de 126 nt de diferencia entre las cepas de la vacuna y la cepa DV de tipo salvaje. Estas mutaciones se acumularon principalmente en el extremo 5′ de la región ORF1 y la región ORF2a (Figura 3).
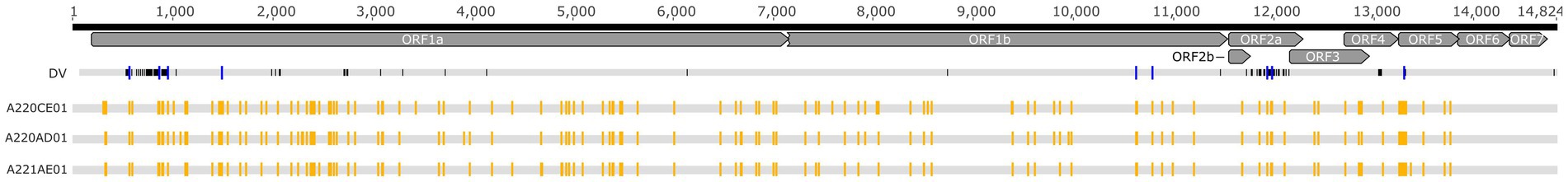 Figura 3. Alineación esquemática de la cepa Porcilis DV y los lotes de vacunas investigados en nuestro estudio. Los cuadrados amarillos muestran la distribución del SNV dentro de los lotes de vacunas, mientras que los cuadrados azules muestran los sitios de SNV que corresponden a las diferencias de nucleótidos entre el DV y los lotes de vacunas.
Figura 3. Alineación esquemática de la cepa Porcilis DV y los lotes de vacunas investigados en nuestro estudio. Los cuadrados amarillos muestran la distribución del SNV dentro de los lotes de vacunas, mientras que los cuadrados azules muestran los sitios de SNV que corresponden a las diferencias de nucleótidos entre el DV y los lotes de vacunas.
A partir de los análisis de secuenciación profunda de amplicones, detectamos una alta complejidad genética. Nuestros análisis de la variabilidad intracepa mostraron que 122, 115 y 113 posiciones SNV estaban presentes en los lotes de Porcilis PRRS de A220CE01, A220AD01 y A221AE01, respectivamente (Figura 3). La mayoría de las variantes menores (un total de 104 sitios) en estos ensamblajes fueron idénticas en los tres lotes de vacunas. La frecuencia media de bases SNV en las variantes menores, calculada a partir de bibliotecas de amplicones duplicadas, varió entre 1,1 y 48,7%, 1 a 47,2% y 1,3 a 48,1%, respectivamente, para los tres lotes (Figura suplementaria S1). La mayoría de los SNV se encontraron en el ORF1a de los tres genomas (55,4 a 60,9%), mientras que no se detectó ningún SNV en las regiones ORF6 y ORF7. El número de SNV en todo el genoma encontrado en nuestros lotes de vacunas fue mucho menor que en algunas cepas de campo, incluida una cepa similar a PRRSV-2 MLV (20, 21). En total, observamos un máximo de 15 nt de diferencia entre nuestros lotes de vacunas y las cepas de Porcilis previamente secuenciadas, y 7 de estas diferencias se identificaron como variantes menores en nuestro análisis de SNV, lo que sugiere que la estructura de cuasiespecies contribuye en parte a la diversidad de viales entre vacunas identificada (Figura suplementaria S2).
La variabilidad intracepa de la vacuna Porcilis MLV se determinó directamente a partir de los viales de la vacuna. De acuerdo con estudios previos, la distribución desigual y la acumulación de SNVs en la región ORF1a es muy típica y es irrelevante si la cepa estudiada es un derivado de la vacuna o una cepa de PRRSV de tipo salvaje (20-22). La ausencia de SNVs en las regiones ORF6 y ORF7 no fue inesperada, ya que estas regiones codificantes están altamente conservadas, a pesar de que algunas cepas de campo albergan espectros mutantes medibles en estos sitios (20-23). Es importante destacar que el patrón de SNV en las cepas de la vacuna Porcilis no se correspondió completamente con los sitios que difieren entre la cepa DV de estos sitios; se repartieron entre ellos un total de 9 variantes menores, seis en ORF1a, dos en ORF2a y una en ORF5 (Figura 3).
4 Conclusión
Los protocolos de estilo ARTIC adecuados para la amplificación de genomas virales completos se han convertido en una piedra angular durante algunos años para describir la diversidad genética y apoyar las investigaciones epidemiológicas moleculares. En nuestro estudio, desarrollamos y evaluamos un método de secuenciación de estilo ARTIC para el PRRSV. Nuestro ensayo fue diseñado para poder evaluar la estabilidad genética de las cepas vacunales de Porcilis MLV, pero anticipamos que el ensayo se puede adaptar fácilmente a otros MLV de PRRSV. Se necesitan más esfuerzos para demostrar la posible aplicación de este protocolo en el campo, ya que los frecuentes eventos de recombinación entre la vacuna y los PRRSV de tipo salvaje en el campo pueden plantear un desafío para el diseño y la implementación exitosos de métodos ARTIC de amplio alcance. Tales saltos en la evolución del genoma viral podrían socavar erróneamente el valor de estos ensayos si no se tienen en cuenta los mecanismos evolutivos comunes al evaluar los datos de secuencia obtenidos.
Declaración de disponibilidad de datos
Los conjuntos de datos presentados en este estudio se pueden encontrar en repositorios en línea. Los nombres de los repositorios y los números de acceso se pueden encontrar en: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA1026488.
Contribuciones de los autores
SJ: Análisis formal, Metodología, Redacción – borrador original, Redacción – revisión y edición. ÁB: Recursos, Supervisión, Redacción – revisión y edición. KC: Metodología, Redacción – revisión y edición. KBl: Análisis formal, Metodología, Redacción – revisión y edición. EK: Análisis formal, Metodología, Redacción – revisión y edición. MD: Metodología, Validación, Redacción – revisión y edición. MH: Recursos, Supervisión, Redacción, Revisión y Edición. KS: Metodología, Supervisión, Redacción – revisión y edición. KBn: Conceptualización, Obtención de fondos, Supervisión, Redacción, revisión y edición.
Financiación
El/los autor/es declara(n) haber recibido apoyo financiero para la investigación, autoría y/o publicación de este artículo. Este trabajo contó con el apoyo del Laboratorio Nacional de Enfermedades Infecciosas de los Animales, Resistencia a los Antimicrobianos, Salud Pública Veterinaria y Seguridad de la Cadena Alimentaria, RRF-2.3.1-21-2022-00001. El trabajo fue elaborado con el apoyo profesional del Programa de Becas para Estudiantes de Doctorado del Programa Cooperativo de Doctorado del Ministerio de Innovación y Tecnología, financiado por el Fondo Nacional de Investigación, Desarrollo e Innovación. Los organismos financiadores no tuvieron ningún papel en el diseño del estudio, en la recopilación, análisis e interpretación de los datos, ni en la redacción del manuscrito.
Conflicto de intereses
MH es empleado de Prophyl Ltd.
El resto de los autores declaran que la investigación se llevó a cabo en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un potencial conflicto de intereses.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o afirmación que pueda ser hecha por su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fvets.2023.1327725/full#supplementary-material
Notas
1. ^https://artic.network/
Referencias
1. Brinton, MA, Gulyaeya, A, Balasuriya, UBR, Dunowska, M, y Faaberg, KS. Perfil de taxonomía de virus ICTV: Arteriviridae 2021. J Gen Virol. (2021) 102:001632. doi: 10.1099/jgv.0.001632
Resumen de PubMed | Texto completo de CrossRef | Google Académico
2. Dokland, T. La biología estructural del PRRSV. Virus Res. (2010) 154:86–97. doi: 10.1016/j.virusres.2010.07.029
Resumen de PubMed | Texto completo de CrossRef | Google Académico
3. Snijder, EJ, Kikkert, M y Fang, Y. Biología molecular y patogénesis del arterivirus. J Gen Virol. (2013) 94:2141–63. doi: 10.1099/vir.0.056341-0
4. Murtaugh, MP, Stadejek, T, Abrahante, JE, Lam, TTY y Leung, FCC. La diversidad cada vez mayor del virus del síndrome respiratorio y reproductivo porcino. Virus Res. (2010) 154:18–30. doi: 10.1016/j.virusres.2010.08.015
Resumen de PubMed | Texto completo de CrossRef | Google Académico
5. Stadejek, T, Stankevicius, A, Murtaugh, MP, y Oleksiewicz, MB. Evolución molecular del PRRSV en Europa: estado actual de la cuestión. Microbiol veterinario. (2013) 165:21–8. doi: 10.1016/j.vetmic.2013.02.029
Resumen de PubMed | Texto completo de CrossRef | Google Académico
6. Chae, C. Vacunas comerciales de virus vivos modificados contra el PRRS. Vacuna. (2021) 9:185. doi: 10.3390/vacunas9020185
Resumen de PubMed | Texto completo de CrossRef | Google Académico
7. Legnardi, M, Cecchinato, M, Homonnay, Z, Dauphin, G y Koutoulis, KC. Variabilidad de la subpoblación viral en diferentes lotes de vacunas contra el virus de la bronquitis infecciosa (IBV) basadas en el linaje GI-23: implicaciones para el campo. Virus Res. (2022) 319:198877. doi: 10.1016/j.virusres.2022.198877
Resumen de PubMed | Texto completo de CrossRef | Google Académico
8. Serrão de Andrade, AA, Soares, AER, Paula de Almeida, LG, y Ciapina, LP. Probar la estabilidad genómica de la cepa de la vacuna brasileña contra la fiebre amarilla utilizando datos de secuenciación de nueva generación. Enfoque de la interfaz. (2021) 11:20200063. doi: 10.1098/rsfs.2020.0063
Resumen de PubMed | Texto completo de CrossRef | Google Académico
9. Eclercy, J, Renson, P, Hirchaud, E, Andraud, M y Beven, V. Evoluciones fenotípicas y genéticas de una vacuna viva modificada para el síndrome respiratorio y reproductivo porcino después de pasajes limitados en cerdos. Vacuna. (2021) 9:392. doi: 10.3390/vacunas9040392
Resumen de PubMed | Texto completo de CrossRef | Google Académico
10. Bøtner, A, Strandbygaard, B, Sørensen, KJ, Have, P, y Madsen, KG. Aparición de síntomas agudos similares al PRRS en rebaños de cerdas después de la vacunación con una vacuna viva modificada contra el PRRS. Rec. Veterinario (1997) 141:497–9. doi: 10.1136/vr.141.19.497
Resumen de PubMed | Texto completo de CrossRef | Google Académico
11. Jiang, YF, Xia, TQ, Zhou, YJ, Yu, LX, Yang, S y Huang, QF. Caracterización de tres aislados de virus del síndrome respiratorio y reproductivo porcino de una sola granja porcina con una fuerte homología con una cepa vacunal. Microbiol veterinario. (2015) 179:242–9. doi: 10.1016/J.VETMIC.2015.06.015
Resumen de PubMed | Texto completo de CrossRef | Google Académico
12. Storgaard, T, Oleksiewicz, M, y Bøtner, A. Examen de las presiones selectivas sobre un virus vivo de la vacuna contra el PRRS. Arch Virol. (1999) 144:2389–401. doi: 10.1007/s007050050652
Resumen de PubMed | Texto completo de CrossRef | Google Académico
13. Wang, J, Zhang, M, Cui, X, Gao, X, Sun, W y Ge, X. El virus del síndrome respiratorio y reproductivo porcino atenuado recupera su virulencia fatal mediante el paso en serie en cerdos o macrófagos alveolares porcinos para aumentar su adaptación a las células diana. Microbiol Spectr. (2022) 10:e0308422. doi: 10.1128/spectrum.03084-22
Resumen de PubMed | Texto completo de CrossRef | Google Académico
14. Zhang, Z, Zhou, L, Ge, X, Guo, X, Han, J y Yang, H. Análisis evolutivo de seis aislados del virus del síndrome respiratorio y reproductivo porcino de una sola granja porcina: virus recombinantes y evolucionados por MLV. Infectar Genet Evol. (2018) 66:111–9. doi: 10.1016/j.meegid.2018.09.024
Resumen de PubMed | Texto completo de CrossRef | Google Académico
15. Bálint, Á, Molnár, T, Kecskeméti, S, Kulcsár, G, Soós, T, y Szabó, PM. Variabilidad genética de las cepas vacunales contra el PRRSV utilizadas en el Programa Nacional de Erradicación. Hungría Vacunas. (2021) 9:849. doi: 10.3390/vacunas9080849
Resumen de PubMed | Texto completo de CrossRef | Google Académico
16. Quick, J, Grubaugh, ND, Pullan, ST, Claro, IM, Smith, AD y Gangavarapu, K. Método de PCR múltiple para la secuenciación MinION e Illumina de genomas de virus del Zika y otros virus directamente a partir de muestras clínicas. Nat Protoc. (2017) 12:1261–76. doi: 10.1038/nprot.2017.066
Resumen de PubMed | Texto completo de CrossRef | Google Académico
17. Gohl, DM, Garbe, J, Grady, P, Daniel, J y Watson, RHB. Un método rápido y rentable de amplicón de cola para secuenciar el SARS-CoV-2. BMC Genómica. (2020) 21:863. doi: 10.1186/s12864-020-07283-6
Resumen de PubMed | Texto completo de CrossRef | Google Académico
18. Balka, G, Hornyák, Á, Bálint, Á, Kiss, I y Kecskeméti, S. Diversidad genética de las cepas del virus del síndrome respiratorio y reproductivo porcino que circulan en las piaras porcinas húngaras. Microbiol veterinario. (2008) 127:128–35. doi: 10.1016/j.vetmic.2007.08.001
Resumen de PubMed | Texto completo de CrossRef | Google Académico
19. Lambisia, AW, Mohammed, KS, Makori TO y Ndwiga, L. Optimización de los cebadores V4 de la red ARTIC del SARS-CoV-2 y el protocolo de secuenciación del genoma completo. Frente Med. (2022) 9:836728. doi: 10.3389/fmed.2022.836728
Resumen de PubMed | Texto completo de CrossRef | Google Académico
20. Brar, MS, Shi, M, Hui, RK-H y Leung, FC-C. Evolución genómica de aislados del virus del síndrome respiratorio y reproductivo porcino (PRRSV) revelados por secuenciación profunda. PLoS Uno. (2014) 9:88807. doi: 10.1371/journal.pone.0088807
Resumen de PubMed | Texto completo de CrossRef | Google Académico
21. Xing, J, Zheng, Z, Cao, X, Wang, Z, Xu, Z, Gao, H, et al. La secuenciación del genoma completo de muestras clínicas revela la diversidad genómica de los virus del síndrome respiratorio y reproductivo porcino que están surgiendo en China. Transbound Emerg Dis. (2022) 69:E2530–40. doi: 10.1111/tbed.14597
Resumen de PubMed | Texto completo de CrossRef | Google Académico
22. Clilverd, H, Martín-Valls, G, Li, Y, Martín, M, Cortey, M, y Mateu, E. Dinámica de la infección, transmisión y evolución después de un brote del virus del síndrome respiratorio y reproductivo porcino. Microbiol frontal. (2023) 14:1109881. doi: 10.3389/fmicb.2023.1109881
Resumen de PubMed | Texto completo de CrossRef | Google Académico
23. Jakab, S, Bali, K, Freytag, C, Pataki, A, Fehér, E y Halas, M. Secuenciación profunda del virus del síndrome respiratorio y reproductivo porcino ORF7: una herramienta prometedora para el diagnóstico y la vigilancia epidemiológica. Animales. (2023) 13:3223. doi: 10.3390/ani13203223
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Palabras clave: virus del síndrome respiratorio y reproductivo porcino, Porcilis MLV, variabilidad genética, variación de nucleótido único, secuenciación profunda, secuenciación de amplicones de mosaico
Cita: Jakab S, Bálint &, Cseri K, Bali K, Kaszab E, Domán M, Halas M, Szarka K y Bányai K (2024) Evaluación de la estabilidad del genoma de la cepa de la vacuna PRRS con un nuevo protocolo de secuenciación de estilo ARTIC. Frente. Vet. Sci. 10:1327725. doi: 10.3389/fvets.2023.1327725
Editado por:
Mengmeng Zhao, Universidad de Foshan, China
Revisado por:
Tong-Qing An, Academia China de Ciencias Agrícolas, China Arruinando a Wang, Universidad de Ganadería y Economía de Henan, China
Derechos de autor © 2024 Jakab, Bálint, Cseri, Bali, Kaszab, Domán, Halas, Szarka y Bányai. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia Creative Commons Attribution License (CC BY).
*Correspondencia: Krisztián Bányai, bkrota@hotmail.com
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o afirmación que pueda hacer su fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía