
Modelos animales en osteosarcoma

Modelos animales en osteosarcoma
 María V. Guijarro*,
María V. Guijarro*,  Steven C. Ghivizzani y
Steven C. Ghivizzani y  C. Parker Gibbs
C. Parker Gibbs- Departamento de Ortopedia y Rehabilitación, Universidad de Florida, Gainesville, FL, EE.UU.
El osteosarcoma (SG) es el tumor óseo primario no hematológico más frecuente en niños y adultos. La quimioterapia citotóxica en dosis altas y la resección quirúrgica han mejorado el pronóstico, con una supervivencia a largo plazo para la enfermedad no metastásica cercana al 70%. Sin embargo, la mayoría de los tumores de SG son de alto grado y tienden a desarrollar rápidamente metástasis pulmonares. A pesar de los avances clínicos, los pacientes con enfermedad metastásica o recaída tienen un mal pronóstico. Para una mejor comprensión de la patogénesis molecular de la SG humana, se han desarrollado varios modelos de ratón con SG modificados genéticamente y se revisarán aquí. Sin embargo, se necesitan mejores modelos animales que recapitulen con mayor precisión la progresión natural de la enfermedad para el desarrollo de marcadores pronósticos y diagnósticos mejorados, así como terapias dirigidas para la SG primaria y metastásica.
Introducción
El osteosarcoma (SG) es una forma altamente maligna de cáncer de hueso caracterizada por la producción de osteoides. Aunque la SG comprende <1 % de los cánceres diagnosticados en los Estados Unidos, es la neoplasia maligna primaria más común del hueso (1, 2). Ocurre predominantemente después de la primera década de vida durante períodos de crecimiento esquelético, con una segunda incidencia máxima en la población de pacientes geriátricos (1, 3). La gran mayoría de la SG en niños, adolescentes y adultos jóvenes es de alto grado y comienza en el espacio intramedular de las ubicaciones metafisarias en los huesos largos de la extremidad inferior. Esto sugiere una relación con las placas de crecimiento activas. Después de una baja incidencia en individuos entre 25 y 59 años de edad, la incidencia de SG aumenta nuevamente en individuos mayores de 60 años de edad, y se asocia con mayor frecuencia con la enfermedad de Paget o la exposición a la radiación (1, 2). Esto puede sugerir que la patogénesis subyacente no es idéntica en pacientes jóvenes y mayores. La SG convencional se presenta en tres subtipos principales según la clasificación histológica: osteoblástica, fibroblástica y condroblástica. El osteoblástico es el más común (alrededor del 60%) con fibroblástico y condroblástico igualmente representados (4).
El osteosarcoma se caracteriza por una invasión local de hueso y tejidos blandos, pérdida de la función de la extremidad afectada y metástasis a distancia, con mayor frecuencia en el pulmón (90%). Las metástasis también se encuentran en el hueso (8-10%) y rara vez en los ganglios linfáticos (5). El tratamiento consiste en la extirpación agresiva del tumor primario para permitir el control local a través de la cirugía conservadora de la extremidad o la amputación. La quimioterapia sistémica (tanto antes como después de la extirpación del tumor) se utiliza para suprimir el desarrollo de metástasis y efectuar la curación. Los regímenes de quimioterapia más comunes comprenden los medicamentos, cisplatino, doxorrubicina y dosis altas de metotrexato en combinación (6–8). Aunque la quimioterapia retrasa el crecimiento del tumor, puede inducir miocardiopatía, pérdida de audición y riesgo de neoplasia maligna secundaria (8, 9). En pacientes sin metástasis en el momento del diagnóstico (80-90%), el tratamiento quirúrgico en combinación con quimioterapia ha dado lugar a tasas de supervivencia a largo plazo que se acercan al 70%. Por el contrario, para los pacientes con metástasis establecidas, actualmente no existe una opción terapéutica confiable para proporcionar control tumoral a largo plazo. A pesar de los intensos esfuerzos para mejorar tanto la quimioterapia como el tratamiento quirúrgico, el 40% de todos los pacientes con SG sucumben a la enfermedad. Específicamente, el resultado clínico para la SG metastásica sigue siendo deficiente; Menos del 30% de los pacientes que presentan metástasis sobreviven 5 años después del diagnóstico inicial. Por lo tanto, existe una necesidad urgente de desarrollar nuevas terapias para los agentes de SG con mayor capacidad para eliminar la carga tumoral sistémica, así como una toxicidad reducida en los tejidos sanos.
Etiología de la SG
El osteosarcoma se caracteriza por un cariotipo complejo y una falta de translocaciones recurrentes. Los enfoques genéticos han identificado varios genes de importancia potencial en el desarrollo y progresión de la enfermedad (10-12). Sin embargo, las alteraciones cromosómicas generalizadas del genoma de la SG han limitado la interpretación de estos hallazgos. Las alteraciones genéticas de la SG suelen ser esporádicas, aunque se ha documentado predisposición genética en pacientes con síndrome de Li-Fraumeni y retinoblastoma. Las deleciones somáticas y las mutaciones puntuales en P53 ocurren en aproximadamente el 50% de la SG humana (13-16) y la mitad de esas mutaciones están asociadas con la pérdida del alelo restante (14). Además, casi el 70% de la SG tiene al menos una alteración del alelo RB (17, 18). Las deleciones homocigotas de RB se observan en el 23% de los tumores, mientras que las mutaciones puntuales aparecen en el 6% (18, 19). Además, también se han reportado numerosas alteraciones que interrumpen la vía RB; por ejemplo, se ha encontrado que la pérdida de función en el locus INK4a/ARF y la amplificación de CDK4 ocurren (uno u otro) en el 22% de la SG (20-22). La prevalencia de estas alteraciones sugeriría que la desregulación del punto de control G1/S y G2/M en el ciclo celular es un evento común en la SG.
Para esto, un tumor de origen desconocido, genética caótica, inicio temprano y comportamiento agresivo, existe la necesidad de modelos más representativos para aprender más sobre la biología de la SG.
Modelos animales en OS
Los modelos animales son muy prometedores para aumentar nuestra comprensión de la base genética de la SG y, lo que es más importante, para avanzar en los estudios preclínicos dirigidos al desarrollo racional de nuevos enfoques terapéuticos, así como a su validación antes de los ensayos clínicos.
Para que cualquier modelo animal de enfermedad humana sea útil e informativo, es preferible recapitular con precisión el curso natural de la enfermedad. Desafortunadamente, la etiología y la patogénesis de la SG no se comprenden completamente; Por lo tanto, el establecimiento y la inducción de modelos experimentales representativos son desafiantes e incompletos. Actualmente, no existe un modelo animal robusto de SG que represente completamente sus características biológicas y clínicas. Lo ideal sería uno en el que hubiera una lesión ósea primaria natural y metástasis pulmonares espontáneas. Hasta la fecha, las principales especies utilizadas para generar modelos de sistemas operativos son el ratón y la rata; sin embargo, la SG que surge en perros también es notable como un modelo validado de SG espontánea.
Muchos aspectos de la biología de la enfermedad se han determinado a partir de una variedad de enfoques de modelos animales. Los modelos de ratón genéticamente modificados de OS han dado al campo mucha información. Sin embargo, la SG espontánea, la SG secundaria como consecuencia de los animales que reciben radiación, las líneas celulares de SG humana y murina y los estudios de xenotrasplante también son importantes para comprender la biología de esta neoplasia maligna.
Modelos caninos
La SG espontánea es mucho más común en perros grandes que en humanos, lo que convierte al perro en un modelo candidato atractivo para estudiar enfermedades humanas (23). La SG canina es indistinguible de los tumores humanos a nivel histológico y de expresión génica (24–27). Las principales diferencias entre los dos son la edad de desarrollo y la prevalencia de la enfermedad. En los perros, la SG es una enfermedad de perros mayores y de razas grandes (6-12 años de edad), y se estima que más de 10,000 casos ocurren anualmente en los Estados Unidos. La mediana del intervalo libre de enfermedad después de la cirugía sola es de 4 meses, y después de la cirugía con quimioterapia, de 13 meses. Esta alta prevalencia y la tasa relativamente rápida de progresión de la enfermedad brindan la oportunidad de modelar el desarrollo y la progresión de la metástasis y evaluar nuevas opciones de tratamiento en un período de tiempo relativamente corto (28-32). Muchos de los genes implicados en la patogénesis de la SG humana parecen participar en la SG canina, incluidos P53, RB y PTEN (33–36).
Aunque la SG canina sirve como un excelente modelo tumoral comparativo para la SG humana, existen algunas limitaciones a considerar. En primer lugar, la SG afecta a los perros geriátricos esqueléticamente maduros, que es diferente de los humanos, donde el pico de incidencia ocurre durante la adolescencia. En segundo lugar, algunas razas tienen mutaciones hereditarias específicas de la línea germinal en ciertos genes que pueden influir en la biología, la progresión y la respuesta al tratamiento de la SG sin impulsar el inicio de la enfermedad (37).
SG secundaria después de la radiación
El desarrollo de modelos de SG para roedores comenzó con la exposición de ratas y ratones a carcinógenos químicos y radiactivos (38-40). Cabe destacar que entre ellos se encuentra el desarrollo de SG en ratas tratadas con P32-ortofosfato, que resultó en una alta incidencia (41). Estos modelos produjeron tumores que se parecían histológicamente al cáncer humano y produjeron líneas celulares que complementan los estudios de SG humana (42). A pesar de la alta penetrancia de los modelos, su relevancia sigue sin estar clara ya que la mayoría de la SG en humanos es esporádica, mientras que el modelo murino inducido por carcinógenos es más representativo de una enfermedad inducida por terapia.
Estudios de xenotrasplante
Existe una cantidad significativa de literatura relacionada con el desarrollo y uso de modelos de xenoinjertos y aloinjertos de células de SG humanas y murinas inyectadas en ratones inmunocomprometidos. Las células inyectadas forman un tumor sólido cultivado localmente en cuestión de días o semanas después de la implantación (42, 43). El uso de estos sistemas se ha convertido en una herramienta destacada en la investigación oncológica actual debido a la rápida aparición, su costo asequible y la facilidad de manejo y mantenimiento. Además, las células derivadas de donantes de OS pueden hacer metástasis a los pulmones, lo que brinda la oportunidad de investigar el crecimiento tumoral primario y secundario. La principal limitación es que el enfoque utiliza células de SG completamente desarrolladas y, por lo tanto, no proporciona información sobre el inicio del tumor y su etiología. Además, dado que el microambiente tumoral puede contribuir significativamente al comportamiento del tumor, tales interacciones pueden perderse al establecer la enfermedad por introducción directa en un animal receptor (44-46). En ciertas circunstancias, la línea celular inyectada puede no ser metastásica en el contexto de roedores, lo que hace imposible estudiar la diseminación de la enfermedad. A pesar de estas limitaciones, muchos grupos han utilizado con éxito este modelo para identificar los factores involucrados en la migración de la SG (47, 48) y, lo que es más importante, para la detección de fármacos con potencial tumoricida (49). Las ventajas distintivas del modelo de inyección de suspensión celular subcutánea son la alta tasa de incidencia y reproducibilidad que permite una titulación precisa del número de células en el inóculo para cuantificar el potencial tumorigénico de las células inyectadas.
Una variación de la inyección de suspensiones celulares en animales receptores es trasplantar piezas de tumor directamente cosechadas del paciente. La ventaja es que las células malignas humanas pueden crecer en su entorno nativo manteniendo la heterogeneidad que puede ser necesaria para su proliferación, que en algunos informes se ha demostrado que mejora el crecimiento tumoral y la metástasis. Con el uso de suspensión celular y trasplantes, las células huésped murinas pueden infiltrarse en el tumor, posiblemente influyendo en las actividades de las células tumorales, y en algunos casos, las células del huésped roedor pueden crecer más de la población de células humanas (50). Alternativamente, se ha demostrado que la implantación ortotópica e intratibial de células de SG induce SG en sitios locales y metastásicos (tibia y pulmón proximales) (43, 51-53). Este enfoque permite el estudio de la formación de tumores primarios dentro de un contexto más nativo, así como las primeras etapas de la progresión metastásica de la SG, reconstituyendo así todo el proceso metastásico. Su uso, sin embargo, está limitado por la falta de reproducibilidad debido en parte a la habilidad técnica requerida para realizar la implantación y la falta asociada de inóculo cuantificable.
Modelos de ratón genéticamente modificados
De los sarcomas con cariotipos complejos, la SG es una de las más estudiadas, como lo demuestra el desarrollo de numerosos modelos de ratón disponibles para esta enfermedad. La capacidad de alterar específicamente la expresión de genes individuales (por pérdida o ganancia de función) estuvo disponible en el ratón con la evolución de las tecnologías de orientación génica (54, 55).
Se han desarrollado muchos modelos murinos de SG para recapitular las mutaciones P53 y RB en la SG humana hereditaria y esporádica. La deleción germinal de P53 resultó en una incidencia de SG del 4% en ratones nulos P53 homocigotos (56) y del 25% en ratones P53 heterocigotos (57), lo que subraya la importancia de P53 alterado en la conducción del sistema operativo. Esta proporción inesperada de formación de tumores, sin embargo, es probable que se deba a la letalidad temprana observada en la población nula homocigótica. Además, el rápido desarrollo, la mayor incidencia de otros tumores (principalmente linfomas) y la larga latencia de la SG (58) requieren el sacrificio de los ratones antes del inicio de la SG, lo que dificulta en muchos casos la utilidad de estos modelos. El papel de P53 se destacó aún más por el análisis tumoral de ratones knock-in P53 que contenían una copia mutante de P53R172H (correspondiente a la mutación del punto caliente R175H en humanos) que no solo desarrollan tumores primarios sino que también hacen metástasis a los pulmones y a otros órganos (59, 60). Por el contrario, los ratones con deleciones de Rb en la línea germinal no desarrollaron SG: la deleción homóloga de Rb es letal embrionaria y los heterocigotos no están predispuestos a la SG (61, 62).
La aplicación de la regulación génica condicional y la disponibilidad de líneas de ratón que expresan Cre específicas del tejido (63) han mejorado enormemente nuestra capacidad para generar modelos específicos de linaje osteogénico mesenquimal que se asemejan más fielmente a la SG humana (55, 64). La mayoría de estos modelos han utilizado la pérdida de P53 con o sin la interrupción de la vía Rb para generar modelos de SG penetrantes (54). Utilizan enfoques de deleción génica condicional restringidos a progenitores mesenquimales multipotentes, osteoblastos comprometidos temprano (preosteoblastos) y la población de osteoblastos (Figura 1) (Tabla 1).
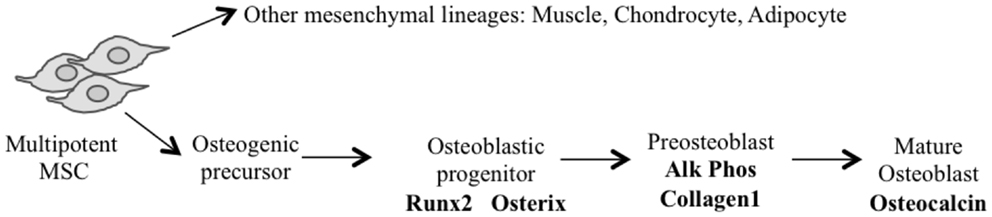 FIGURA 1. SE MUESTRA EL MODELO DE DIFERENCIACIÓN DE OSTEOBLASTOS Y LA ETAPA PUTATIVA DE EXPRESIÓN DE CRE.
FIGURA 1. SE MUESTRA EL MODELO DE DIFERENCIACIÓN DE OSTEOBLASTOS Y LA ETAPA PUTATIVA DE EXPRESIÓN DE CRE.
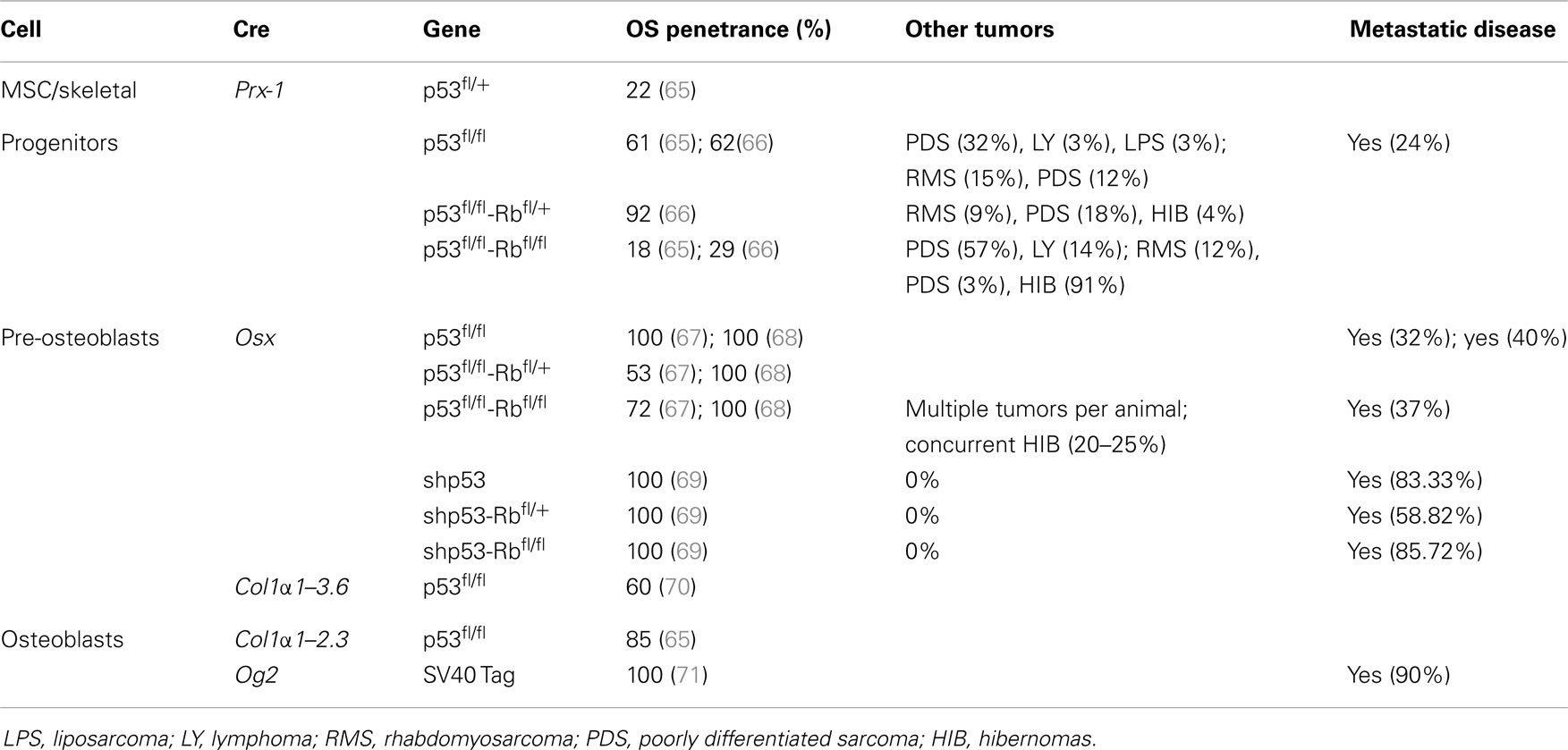 TABLA 1. RESUMEN DE MODELOS MURINOS DE SG MODIFICADOS GENÉTICAMENTE.
TABLA 1. RESUMEN DE MODELOS MURINOS DE SG MODIFICADOS GENÉTICAMENTE.
Usando Cre recombinasa activada por el promotor del gen Paired related homebox 1 (Prx1-Cre) (72) que elimina los alelos flanqueados por LoxP en el mesénquima temprano de las extremidades (células multipotenciales), el 22% de los ratones con heterocigosidad mediada por P53 desarrollaron SG. No es sorprendente que la deleción homocigótica de P53 tuviera un aumento de tres veces en la incidencia de SG sobre los animales heterocigotos. En contraste, la deleción de Rb en los progenitores mesenquimales que expresan Prx no produjo ningún tumor de SG (65, 66). Curiosamente, la mayor incidencia (92%) de SG ocurrió con la deleción combinada de un alelo de Rb con la deleción homocigótica P53 (66). La deleción homocigótica de ambos genes dio lugar a una mayor formación de tumores inespecíficos con solo 18 % de tumores con SG y el resto siendo sarcomas de tejidos blandos poco diferenciados (PD-STS) y linfoma (65, 66).
Para una deleción más restringida de genes en el linaje osteoblasto, se han utilizado promotores de genes que van desde los expresados temprano en el compromiso de progenitores como Osterix 1 y Colágeno1 α1-3.6 hasta los expresados en precursores de osteoblastos más restringidos por linaje como Colágeno1 α1-2.3 y osteocalcina (Og2). Se ha observado el desarrollo de SG con una penetrancia del 100% (67, 68) después de la deleción específica de osteoblastos de P53 utilizando la expresión Cre mediada por Osterix (Osx-Cre) (73). Al igual que con los progenitores mesenquimales, las deleciones de Rb no tienen efecto y la deleción combinada de Rb y P53 en osteoblastos volvió a generar SG fibroblástica o indiferenciada con alta penetrancia (100%) (67, 68). La utilidad traslacional potencial es la existencia de SG metastásica espontánea de baja latencia similar a los tumores humanos en la que las células se detienen en su diferenciación (67, 68). Aunque la mayor proporción de tumores fue la SG cuando P53 se eliminó condicionalmente, también se informó que se generaron tumores neuroendocrinos e hibernomas en varios ratones (67, 68). Sin embargo, Walkley et al. enriquecieron el fondo C57BL/6 de la cepa de ratón y el porcentaje de hibernomas se redujo, sugiriendo un posible impacto de las cepas de ratón en el fenotipo observado (69). Un estudio reciente en ratones que expresaron el antígeno SV40 T/t (Tag) en osteoblastos maduros bajo el Og2 (74) mostró SG con penetrancia completa (71) y 90% de incidencia de metástasis pulmonares. Un análisis adicional de los tumores derivados de este modelo reveló una deleción genómica recurrente del gen Prkar1a en un subconjunto específico también en la SG humana. El ARN shtransgénico se ha utilizado para derribar específicamente P53 (en lugar de eliminar) utilizando el transgén Osx-Cre (69). Estos ratones desarrollan SG osteoblástica con una penetrancia del 100%, y aunque tienen una latencia más larga hasta el inicio del tumor, con mayor frecuencia se desarrollan en huesos largos y son altamente metastásicos (pulmón e hígado), características similares a la SG humana. Este modelo no ha desarrollado ningún tumor que no sea SG.
Independientemente de la etapa de desarrollo en la que Cre se activa, la latencia del sistema operativo es esencialmente la misma cuando se compara P53 solo o en combinación con Rb. El uso de Cre en células más primitivas (Prx), sin embargo, conduce al desarrollo de tumores de otros linajes mesenquimales con mayor frecuencia.
Posiblemente proporcionando información sobre los eventos iniciadores de la SG (70), una característica celular prominente de la inactivación condicional de P53 en progenitores osteoblásticos es la hiperproliferación de osteoblastos antes de la formación del tumor. Se ha propuesto que Rb tiene un papel en la influencia de la diferenciación tardía de osteoblastos al interactuar con Runx2 (75). Sin embargo, varios estudios independientes han demostrado que la eliminación de Rb sola no es suficiente para inducir SG. Los diferentes enfoques experimentales sugieren fuertemente que la mutación en la vía p53 puede servir como un evento iniciador en la SG, con una mutación posterior en la vía Rb que acelera fuertemente el desarrollo tumoral.
Estos modelos de ratón diseñados de SG reproducen muchas características de la SG humana, incluidas firmas similares de transcripción de genes (76) y complejidad citogenética. Sin embargo, los sitios de formación de tumores primarios en ratones Cre-loxP no recapitulan la enfermedad humana espontánea. La mayoría de las lesiones (85%) surgen en sitios esqueléticos axiales (mandibulo, maxilar, costilla / vértebra, cráneo, esternón), mientras que en el 13,6% de los tumores se desarrollaron a partir del esqueleto apendicular (pata trasera, pata delantera) (68). Esto contrasta con la distribución anatómica de la SG diagnosticada en humanos, siendo el fémur distal, la tibia proximal y el húmero proximal los sitios más comunes involucrados y solo el 10% se desarrolla en el esqueleto axial, más comúnmente la pelvis (5). Sólo en un estudio (69) el tumor surgió principalmente en los huesos largos. Además, la frecuencia observada de metástasis a distancia fue comparativamente baja en comparación con la enfermedad humana, excepto por el modelo de derribo P53 (69). A diferencia de una deleción completa de P53, las células tumorales primarias proliferaron más lentamente y los animales no tuvieron que ser sacrificados por el tamaño del tumor local antes de completar el proceso metastásico. Además, el sitio primario de metástasis en la SG humana es predominantemente el parénquima pulmonar, mientras que en ratones Cre-loxP, los sitios de metástasis fueron más diversos, y tanto el pulmón como el hígado se vieron afectados en proporciones casi iguales.
Otros genes como C-FOS (77, 78), TWIST (79), p14ARF (80), p16INK4a (81), PRKAR1A (71) y p21CIP (82) también se han implicado en la patogénesis de la SG basada en estudios de muestras de SG humanas. Su mutación parece complementar los defectos en las vías P53 y RB, y su participación en la osteosarcomagénesis también se demuestra a partir de modelos de ratón genéticamente modificados. Proporcionan información importante sobre la genética de la SG, pero la larga latencia combinada con la baja penetrancia hace que la utilización de estos modelos sea menos práctica.
Terapias dirigidas en la SG
El osteosarcoma es muy resistente a la terapia y, por lo tanto, existe una necesidad urgente de tratar eficazmente a los pacientes afectados. La aparición de nuevos fármacos contra el cáncer y el pequeño número de pacientes elegibles para ensayos clínicos de fase temprana presentan otro desafío en las pruebas clínicas de nuevos compuestos para el tratamiento de la SG. Como se discutió anteriormente, los modelos de xenotrasplante han proporcionado la mayor utilidad para el cribado preclínico de fármacos con potencial tumoricida. Con este fin, el Instituto Nacional del Cáncer (NCI) ha implementado el Programa de Pruebas Preclínicas Pediátricas (PPTP), un consorcio de instituciones en los Estados Unidos y en Australia. Su objetivo es identificar agentes con actividad significativa en paneles de modelos de xenoinjertos de ratón que representan los cánceres pediátricos más comunes, incluida la SG (83). El programa ha sido exitoso, lo que ha llevado a ensayos clínicos de fase I y II para cixutumumab, sorafenib y rapamicina para el tratamiento de la SG. (84–86). En cada caso, estos agentes demostraron altos niveles de respuesta en el PPTP y fueron bien tolerados con actividad antitumoral prometedora en algunos pacientes adultos y pediátricos.
El uso de modelos de SG espontáneos y transgénicos para el cribado de alto rendimiento de fármacos anti-SG se ve obstaculizado debido a consideraciones prácticas asociadas con el costo y el tiempo de generación de un número suficiente de animales para obtener datos estadísticamente significativos. Esto se debe a las variaciones en el inicio de la enfermedad, así como a la heterogeneidad, incidencia y progresión del tumor. Sin embargo, la reciente generación de animales transgénicos que expresan ARNshpara derribar P53 (69) representa un avance potencial con respecto al cribado preclínico. A diferencia de los enfoques convencionales de deleción génica mediada por Cre, los ratones derribados P53 exhibieron una penetrancia del 100% para la SG osteoblástica (la forma más común de la enfermedad). Además, los tumores estaban presentes con mayor frecuencia en los huesos largos y se diseminaban preferentemente a los pulmones, de acuerdo con la SG humana.
Otra consideración para las pruebas preclínicas en modelos in vivo es la medición precisa de la carga de la enfermedad en sitios no accesibles. El uso de imágenes in vivo ofrece la oportunidad de detectar y monitorear el desarrollo y la progresión de la enfermedad. Sin embargo, los sistemas de imágenes son costosos y no siempre son ampliamente accesibles para muchos investigadores. La SG tiene la ventaja de que el tumor primario en modelos de ratón genéticamente modificados aparece en los huesos largos y, por lo tanto, es más accesible que los tumores abdominales. El monitoreo/visualización de micrometástasis representa un mayor desafío debido a su pequeño tamaño. La evaluación inexacta de la diseminación metastásica en estudios preclínicos puede conducir a resultados decepcionantes en los ensayos clínicos. En consecuencia, existe un gran interés en refinar los métodos para permitir la detección reproducible y ultrasensible de metástasis a nivel de una sola célula. Por lo tanto, el enfoque principal está en las técnicas que permiten la detección de células tumorales in vivo, como la tomografía microcomputarizada (micro-CT), la tomografía por emisión de positrones (PET), la bioluminiscencia o la imagen de fluorescencia.
Conclusión
Nuestra comprensión de la biología de la SG humana se ve obstaculizada por su rápido inicio, baja prevalencia y ausencia de condiciones predisponentes o lesiones precursoras. Con tejido humano limitado disponible para el estudio, los modelos animales proporcionan una herramienta valiosa para investigar los mecanismos subyacentes que impulsan la iniciación tumoral, la progresión, los eventos metastásicos y las intervenciones terapéuticas. Si bien estos modelos aún tienen que recapitular fielmente todos los aspectos de la SG, no hay duda de que el estudio de los modelos animales de SG ha permitido comprender la genética de la iniciación tumoral, así como los perfiles celulares y moleculares del crecimiento tumoral y la metástasis. En particular, los estudios de eliminación de genes han sido fundamentales para identificar mutaciones genéticas que promueven el inicio del tumor de SG (P53), así como mutaciones cooperativas que aumentan la incidencia de la enfermedad (RB, c-FOS).
Con el uso de marcadores específicos de linaje celular, ahora es posible introducir mutaciones genéticas mediante la orientación secuencial desde precursores tempranos (células mesenquimales multipotentes) hasta células osteoblásticas más maduras (osteoblastos a osteocitos) para investigar la incidencia de SG y la patología tumoral. Con esta estrategia, Prx1 y Osx se han utilizado para identificar células mesenquimales y osteoprogenitoras, respectivamente, después de la mutación condicional de P53. Queda por ver, sin embargo, si estas poblaciones son realmente distintas, ya que Prx1 podría coexpresarse con Osx en una cierta subpoblación de células. Otra consideración particularmente relevante en la SG es su heterogeneidad tumoral entre los pacientes, lo que sugiere que múltiples tipos celulares podrían actuar como células de origen. Además, este concepto de heterogeneidad pone en tela de juicio la utilidad de los modelos que explotan la manipulación de un solo gen. Su consideración puede permitir un análisis más sistemático de las lesiones genéticas implicadas en el inicio y la progresión de la SG y podría servir como plataforma para la identificación de biomarcadores tempranos de la enfermedad. La identificación de células de origen también puede tener implicaciones importantes en la prevención de la recaída y dilucidar vías moleculares clave y mutaciones impulsoras que podrían conducir a nuevos enfoques terapéuticos para prevenir la enfermedad.
Por lo tanto, aunque por ahora, los modelos convencionales de trasplante ortotópico y subcutáneo seguirán siendo indispensables para continuar el estudio de la SG in vivo, es necesario desarrollar nuevos modelos de SG espontánea para ampliar nuestra comprensión de la biología de la SG. Los modelos que reproducen con precisión el establecimiento de micrometástasis espontáneas son necesarios para investigar nuevos agentes antimetastásicos, ya que este escenario clínico es con mayor frecuencia el evento letal para los pacientes con esta forma de cáncer.
Declaración de conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un posible conflicto de intereses.
Referencias
1. Mirabello L, Troisi RJ, Savage SA. Patrones internacionales de incidencia de osteosarcoma en niños y adolescentes, mediana edad y ancianos. Int J Cancer (2009) 125(1):229–34. doi: 10.1002/ijc.24320
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
2. Mirabello L, Troisi RJ, Savage SA. Tasas de incidencia y supervivencia del osteosarcoma de 1973 a 2004: datos del Programa de Vigilancia, Epidemiología y Resultados Finales. Cáncer (2009) 115(7):1531–43. doi:10.1002/cncr.24121
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
3. McKenna RJ, Schwinn CP, Higinbotham NL. Sarcoma osteogénico en niños. CA Cancer J Clin (1966) 16(1):26–8.
4. Gorlick R. Conceptos actuales sobre la biología molecular del osteosarcoma. Cancer Treat Res (2009) 152:467–78. doi:10.1007/978-1-4419-0284-9_27
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
5. Luetke A, Meyers PA, Lewis I, Juergens H. Tratamiento del osteosarcoma: ¿dónde estamos? Una revisión de vanguardia. Cancer Treat Rev (2014) 40(4):523–32. doi:10.1016/j.ctrv.2013.11.006
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
6. Janeway KA, Grier HE. Secuelas de la terapia médica con osteosarcoma: una revisión de toxicidades agudas raras y efectos tardíos. Lancet Oncol (2010) 11(7):670–8. doi:10.1016/S1470-2045(10)70062-0
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
7. Picci P, Ferrari S, Bacci G, Gherlinzoni F. Recomendaciones de tratamiento para el osteosarcoma y sarcomas de tejidos blandos en adultos. Drugs (1994) 47(1):82–92. doi:10.2165/00003495-199447010-00006
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
8. Sloet van Oldruitenborgh-Oosterbaan MM, Klein WR, Misdorp W. Diagnóstico diferencial de parches ‘fúngicos’ no curativos en caballos. Tijdschr Diergeneeskd (1994) 119(24):756–9.
9. Savage SA, Mirabello L, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, et al. El estudio de asociación de todo el genoma identifica dos loci de susceptibilidad para el osteosarcoma. Nat Genet (2013) 45(7):799–803. doi:10.1038/ng.2645
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
10. Sandberg AA, Puente JA. Actualizaciones sobre la citogenética y genética molecular de tumores óseos y de tejidos blandos: osteosarcoma y tumores relacionados. Cancer Genet Cytogenet (2003) 145(1):1–30. doi:10.1016/S0165-4608(02)00848-8
11. Pasic I, Shlien A, Durbin AD, Stavropoulos DJ, Baskin B, Ray PN, et al. Los cambios recurrentes en el número focal de copias y la pérdida de heterocigosidad implican dos ARN no codificantes y un gen supresor de tumores en el cromosoma 3q13.31 en el osteosarcoma. Cancer Res (2010) 70(1):160–71. doi:10.1158/0008-5472.CAN-09-1902
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
12. Fletcher JA, Gebhardt MC, Kozakewich HP. Aberraciones citogenéticas en osteosarcomas. Deleciones no aleatorias, anillos y cromosomas de doble minuto. Cancer Genet Cytogenet (1994) 77(1):81–8. doi:10.1016/0165-4608(94)90154-6
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
13. Overholtzer M, Rao PH, Favis R, Lu XY, Elowitz MB, Barany F, et al. La presencia de mutaciones p53 en osteosarcomas humanos se correlaciona con altos niveles de inestabilidad genómica. Proc Natl Acad Sci U S A (2003) 100(20):11547–52. doi:10.1073/pnas.1934852100
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
14. Miller CW, Aslo A, Tsay C, Slamon D, Ishizaki K, Toguchida J, et al. Frecuencia y estructura de los reordenamientos de p53 en osteosarcoma humano. Cancer Res (1990) 50(24):7950–4.
15. Miller CW, Aslo A, Won A, Tan M, Lampkin B, Koeffler HP. Alteraciones de los genes p53, Rb y MDM2 en osteosarcoma. J Cancer Res Clin Oncol (1996) 122(9):559–65. doi:10.1007/BF01213553
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
16. Toguchida J, Yamaguchi T, Ritchie B, Beauchamp RL, Dayton SH, Herrera GE, et al. Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res (1992) 52(22):6194–9.
17. Hansen MF, Koufos A, Gallie BL, Phillips RA, Fodstad O, Brogger A, et al. Osteosarcoma and retinoblastoma: a shared chromosomal mechanism revealing recessive predisposition. Proc Natl Acad Sci U S A (1985) 82(18):6216–20. doi:10.1073/pnas.82.18.6216
18. Wadayama B, Toguchida J, Shimizu T, Ishizaki K, Sasaki MS, Kotoura Y, et al. Mutation spectrum of the retinoblastoma gene in osteosarcomas. Cancer Res (1994) 54(11):3042–8.
19. Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature (1986) 323(6089):643–6. doi:10.1038/323643a0
20. Maitra A, Roberts H, Weinberg AG, Geradts J. Loss of p16(INK4a) expression correlates with decreased survival in pediatric osteosarcomas. Int J Cancer (2001) 95(1):34–8. doi:10.1002/1097-0215(20010120)95:1<34::AID-IJC1006>3.0.CO;2-V
21. Miller CW, Aslo A, Campbell MJ, Kawamata N, Lampkin BC, Koeffler HP. Alterations of the p15, p16, and p18 genes in osteosarcoma. Cancer Genet Cytogenet (1996) 86(2):136–42. doi:10.1016/0165-4608(95)00216-2
22. Wei G, Lonardo F, Ueda T, Kim T, Huvos AG, Healey JH, et al. CDK4 gene amplification in osteosarcoma: reciprocal relationship with INK4A gene alterations and mapping of 12q13 amplicons. Int J Cancer (1999) 80(2):199–204. doi:10.1002/(SICI)1097-0215(19990118)80:2<199::AID-IJC7>3.0.CO;2-4
23. Misdorp W. Skeletal osteosarcoma. Animal model: canine osteosarcoma. Am J Pathol (1980) 98(1):285–8.
24. Selvarajah GT, Kirpensteijn J, van Wolferen ME, Rao NA, Fieten H, Mol JA. Gene expression profiling of canine osteosarcoma reveals genes associated with short and long survival times. Mol Cancer (2009) 8:72. doi:10.1186/1476-4598-8-72
25. Paoloni M, Davis S, Lana S, Withrow S, Sangiorgi L, Picci P, et al. La genómica de especies cruzadas de tumores caninos descubre objetivos relacionados con la progresión del osteosarcoma. BMC Genómica (2009) 10:625. doi:10.1186/1471-2164-10-625
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
26. Mueller F, Fuchs B, Kaser-Hotz B. Comparative biology of human and canine osteosarcoma. Anticancer Res (2007) 27(1A):155–64.
27. Rankin KS, Starkey M, Lunec J, Gerrand CH, Murphy S, Biswas S. De perros y hombres: la biología comparada como herramienta para el descubrimiento de nuevos biomarcadores y dianas de desarrollo de fármacos en osteosarcoma. Pediatr Blood Cancer (2012) 58(3):327–33. doi:10.1002/pbc.23341
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
28. Kirpensteijn J, Teske E, Kik M, Klenner T, Rutteman GR. Lobaplatin as an adjuvant chemotherapy to surgery in canine appendicular osteosarcoma: a phase II evaluation. Anticancer Res (2002) 22(5):2765–70.
29. Kirpensteijn J, Timmermans-Sprang EP, van Garderen E, Rutteman GR, Lantinga-van Leeuwen IS, Mol JA. Expresión génica de la hormona del crecimiento en placas de crecimiento canino normal y osteosarcoma espontáneo. Mol Cell Endocrinol (2002) 197(1–2):179–85. doi:10.1016/S0303-7207(02)00269-1
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
30. Kirpensteijn J, Kik M, Rutteman GR, Teske E. Prognostic significance of a new histologic grading system for canine osteosarcoma. Vet Pathol (2002) 39(2):240–6. doi:10.1354/vp.39-2-240
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
31. Lascelles BD, Dernell WS, Correa MT, Lafferty M, Devitt CM, Kuntz CA, et al. Mejora de la supervivencia asociada con la infección postoperatoria de la herida en perros tratados con cirugía de salvamento de extremidades para el osteosarcoma. Ann Surg Oncol (2005) 12(12):1073–83. doi:10.1245/ASO.2005.01.011
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
32. Kirpensteijn J, Kik M, Teske E, Rutteman GR. TP53 gene mutations in canine osteosarcoma. Vet Surg (2008) 37(5):454–60. doi:10.1111/j.1532-950X.2008.00407.x
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
33. van Leeuwen IS, Cornelisse CJ, Misdorp W, Goedegebuure SA, Kirpensteijn J, Rutteman GR. P53 gene mutations in osteosarcomas in the dog. Cancer Lett (1997) 111(1–2):173–8. doi:10.1016/S0304-3835(96)04529-6
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
34. Johnson AS, Couto CG, Weghorst CM. Mutación del gen supresor de tumores p53 en osteosarcomas de aparición espontánea del perro. Carcinogénesis (1998) 19(1):213–7. doi:10.1093/carcin/19.1.213
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
35. Levine RA, Fleischli MA. Inactivación de las vías familiares de p53 y retinoblastoma en líneas celulares de osteosarcoma canino. Vet Pathol (2000) 37(1):54–61. doi:10.1354/vp.37-1-54
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
36. Levine RA, Forest T, Smith C. El supresor tumoral PTEN está mutado en líneas celulares y tumores de osteosarcoma canino. Vet Pathol (2002) 39(3):372–8. doi:10.1354/vp.39-3-372
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
37. Liao AT, McMahon M, London CA. Identification of a novel germinline MET mutation in dogs. Anim Genet (2006) 37(3):248–52. doi:10.1111/j.1365-2052.2006.01415.x
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
38. Martin TJ, Ingleton PM, Underwood JC, Michelangeli VP, Hunt NH, Melick RA. Adenilato ciclasa sensible a la hormona paratiroidea en sarcoma de rata osteogénico trasplantable inducido. Nature (1976) 260(5550):436–8. doi:10.1038/260436a0
39. Ingleton PM, Coulton LA, Preston CJ, Martin TJ. Fosfatasa alcalina en suero y tumor de ratas portadoras de un sarcoma osteogénico trasplantable sensible a hormonas. Eur J Cancer (1979) 15(5):685–91. doi:10.1016/0014-2964(79)90142-7
40. Underwood JC, Melick RA, Loomes RS, Dangerfield VM, Crawford A, Coulton L, et al. Correlaciones estructurales y funcionales en sarcomas osteogénicos trasplantables sensibles a la hormona paratiroidea. Eur J Cancer (1979) 15(9):1151–8. doi:10.1016/0014-2964(79)90131-2
41. Bensted JP, Blackett NM, Lamerton LF. Estudios sobre el desarrollo de tumores óseos inducidos por radiación. Acta Unio Int Contra Cancrum (1959) 15:559–60.
42. Ek ET, Dass CR, Choong PF. Modelos de ratón comúnmente utilizados de osteosarcoma. Crit Rev Oncol Hematol (2006) 60(1):1–8. doi:10.1016/j.critrevonc.2006.03.006
43. Dass CR, Ek ET, Choong PF. Human xenograft osteosarcoma models with spontaneous metastasis in ratones: clinical relevance and applicability for drug testing. J Cancer Res Clin Oncol (2007) 133(3):193–8. doi:10.1007/s00432-006-0157-x
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
44. Becher OJ, Holland EC. Los modelos modificados genéticamente tienen ventajas sobre los xenoinjertos para estudios preclínicos. Cancer Res (2006) 66(7):3355–8. doi:10.1158/0008-5472.CAN-05-3827 discusión 8-9,
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
45. Sharpless NE, Depinho RA. El ratón poderoso: modelos de ratón genéticamente modificados en el desarrollo de medicamentos contra el cáncer. Nat Rev Drug Discov (2006) 5(9):741–54. doi:10.1038/nrd2110
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
46. Frese KK, Tuveson DA. Maximización de los modelos de cáncer de ratón. Nat Rev Cancer (2007) 7(9):645–58. doi:10.1038/NRC2192
47. Khanna C, Prehn J, Yeung C, Caylor J, Tsokos M, Helman L. Un modelo ortotópico de osteosarcoma murino con variantes relacionadas clonalmente que difieren en el potencial metastásico pulmonar. Clin Exp Metastasis (2000) 18(3):261–71. doi:10.1023/A:1006767007547
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
48. Khanna C, Khan J, Nguyen P, Prehn J, Caylor J, Yeung C, et al. Diferencias asociadas a metástasis en la expresión génica en un modelo murino de osteosarcoma. Cancer Res (2001) 61(9):3750–9.
49. Sampson VB, Gorlick R, Kamara D, Anders Kolb E. Una revisión de las terapias dirigidas evaluadas por el programa de pruebas preclínicas pediátricas para el osteosarcoma. Frente Oncol (2013) 3:132. doi:10.3389/fonc.2013.00132
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
50. Kresse SH, Meza-Zepeda LA, Machado I, Llombart-Bosch A, Myklebost O. Los modelos preclínicos de xenoinjerto de sarcoma humano muestran pérdida no aleatoria de aberraciones. Cancer (2012) 118(2):558–70. doi:10.1002/cncr.26276
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
51. Berlin O, Samid D, Donthineni-Rao R, Akeson W, Amiel D, Woods VL Jr. Development of a novel spontaneous metastasis model of human osteosarcoma transplanted orthotopically into bone of athymic ratones. Cancer Res (1993) 53(20):4890–5.
52. Crnalic S, Hakansson I, Boquist L, Lofvenberg R, Brostrom LA. Un nuevo modelo de metástasis espontánea de osteosarcoma humano desarrollado mediante trasplante ortotópico de tejido tumoral intacto en tibia de ratones desnudos. Clin Exp Metastasis (1997) 15(2):164–72. doi:10.1023/A:1018456911823
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
53. Yuan J, Ossendorf C, Szatkowski JP, Bronk JT, Maran A, Yaszemski M, et al. Los osteosarcomas humanos osteoblásticos y osteolíticos se pueden estudiar con un nuevo modelo de ratón xenoinjerto que produce metástasis espontáneas. Cancer Invest (2009) 27(4):435–42. doi:10.1080/07357900802491477
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
54. Janeway KA, Walkley CR. Modelado del osteosarcoma humano en el ratón: de la cabecera al banco. Hueso (2010) 47(5):859–65. doi:10.1016/j.bone.2010.07.028
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
55. Ng AJ, Mutsaers AJ, Baker EK, Walkley CR. Modelos de ratón genéticamente modificados y osteosarcoma humano. Clin Sarcoma Res (2012) 2(1):19. doi:10.1186/2045-3329-2-19
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
56. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, et al. Los ratones deficientes para p53 son normales en el desarrollo pero susceptibles a tumores espontáneos. Nature (1992) 356(6366):215–21. doi:10.1038/356215a0
57. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Análisis del espectro tumoral en ratones mutantes p53. Curr Biol (1994) 4(1):1–7. doi:10.1016/S0960-9822(00)00002-6
58. Lavigueur A, Maltby V, Mock D, Rossant J, Pawson T, Bernstein A. Alta incidencia de tumores de pulmón, hueso y linfoide en ratones transgénicos que sobreexpresan alelos mutantes del oncogén p53. Mol Cell Biol (1989) 9(9):3982–91.
59. Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Ganancia de función de una mutación de punto caliente p53 en un modelo de ratón del síndrome de Li-Fraumeni. Cell (2004) 119(6):861–72. doi:10.1016/j.cell.2004.11.006
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
60. Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Ganancia de función de p53 mutante en dos modelos de ratón del síndrome de Li-Fraumeni. Cell (2004) 119(6):847–60. doi:10.1016/j.cell.2004.11.004
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
61. Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Efectos tumorigénicos cooperativos de mutaciones de la línea germinal en Rb y p53. Nat Genet (1994) 7(4):480–4. doi:10.1038/ng0894-480
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
62. Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, et al. Los ratones deficientes para Rb son inviables y muestran defectos en neurogénesis y hematopoyesis. Nature (1992) 359(6393):288–94. doi:10.1038/359288a0
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
63. VanKoevering KK, Williams BO. Cepas de ratones transgénicos para la eliminación condicional de genes durante el desarrollo esquelético. IBMS boneKEy (2008) 5:151–70. doi:10.1138/20080312
64. Mohseny AB, Hogendoorn PC, Cleton-Jansen AM. Modelos de osteosarcoma: desde líneas celulares hasta pez cebra. Sarcoma (2012) 2012:417271. doi:10.1155/2012/417271
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
65. Lin PP, Pandey MK, Jin F, Raymond AK, Akiyama H, Lozano G. Mutación dirigida de p53 y Rb en células mesenquimales de la yema de la extremidad produce sarcomas en ratones. Carcinogénesis (2009) 30(10):1789–95. doi:10.1093/carcin/bgp180
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
66. Calo E, Quintero-Estades JA, Danielian PS, Nedelcu S, Berman SD, Lees JA. Rb regula la elección del destino y el compromiso de linaje in vivo. Nature (2010) 466(7310):1110–4. doi:10.1038/nature09264
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
67. Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, et al. Osteosarcoma metastásico inducido por inactivación de Rb y p53 en el linaje osteoblasto. Proc Natl Acad Sci U S A (2008) 105(33):11851–6. doi:10.1073/pnas.0805462105
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
68. Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, et al. El osteosarcoma condicional de ratón, dependiente de la pérdida de p53 y potenciado por la pérdida de Rb, imita la enfermedad humana. Genes Dev (2008) 22(12):1662–76. doi:10.1101/gad.1656808
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
69. Mutsaers AJ, Ng AJ, Baker EK, Russell MR, Chalk AM, Wall M, et al. Modelado de distintos subtipos de osteosarcoma in vivo utilizando Cre:lox y shRNA transgénico restringido por linaje. Bone (2013) 55(1):166–78. doi:10.1016/j.bone.2013.02.016
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
70. Lengner CJ, Steinman HA, Gagnon J, Smith TW, Henderson JE, Kream BE, et al. La diferenciación osteoblástica y el desarrollo esquelético están regulados por la señalización de Mdm2-p53. J Cell Biol (2006) 172(6):909–21. doi:10.1083/jcb.200508130
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
71. Molyneux SD, Di Grappa MA, Beristain AG, McKee TD, Wai DH, Paderova J, et al. Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclass in mice. J Clin Invest (2010) 120(9):3310–25. doi:10.1172/JCI42391
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
72. Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expresión de Cre recombinasa en el brote de la extremidad del ratón en desarrollo impulsado por un potenciador Prxl. Genesis (2002) 33(2):77–80. doi:10.1002/gene.10092
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
73. Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Desarrollo (2006) 133(16):3231–44. doi:10.1242/dev.02480
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
74. Frendo JL, Xiao G, Fuchs S, Franceschi RT, Karsenty G, Ducy P. Functional hierarchy between two OSE2 elements in the control of osteocalcin gene expression in vivo. J Biol Chem (1998) 273(46):30509–16. doi:10.1074/jbc.273.46.30509
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
75. Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, et al. La proteína del retinoblastoma actúa como un coactivador transcripcional necesario para la diferenciación osteogénica. Mol Cell (2001) 8(2):303–16. doi:10.1016/S1097-2765(01)00327-6
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
76. Kuijjer ML, Namlos HM, Hauben EI, Machado I, Kresse SH, Serra M, et al. Los perfiles de expresión de ARNm del osteosarcoma central primario de alto grado se conservan en líneas celulares y xenoinjertos. BMC Med Genomics (2011) 4:66. doi:10.1186/1755-8794-4-66
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
77. Weisstein JS, Majeska RJ, Klein MJ, Einhorn TA. Detección de la expresión de c-fos en lesiones musculoesqueléticas benignas y malignas. J Orthop Res (2001) 19(3):339–45. doi:10.1016/S0736-0266(00)90020-2
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
78. Ruther U, Komitowski D, Schubert FR, Wagner EF. La expresión de C-Fos induce tumores óseos en ratones transgénicos. Oncogén (1989) 4(7):861–5.
79. Entz-Werle N, Choquet P, Neuville A, Kuchler-Bopp S, Clauss F, Danse JM, et al. Ratones doble mutantes dirigidos a la derivación: un nuevo modelo de osteosarcoma espontáneo que imita la enfermedad humana. Transl Oncol (2010) 3(6):344–53. doi:10.1593/tlo.10169
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
80. Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, Zevenhoven J, et al. p15Ink4b es un supresor tumoral crítico en ausencia de p16Ink4a. Nature (2007) 448(7156):943–6. doi:10.1038/nature06084
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
81. Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, et al. La pérdida de p16Ink4a con retención de p19Arf predispone a los ratones a la tumorigénesis. Nature (2001) 413(6851):86–91. doi:10.1038/35092592
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
82. Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient ratones. Cancer Res (2001) 61(16):6234–8.
83. Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. El programa de pruebas preclínicas pediátricas: descripción de modelos y resultados de pruebas tempranas. Pediatr Blood Cancer (2007) 49(7):928–40. doi:10.1002/pbc.21078
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
84. Chawla SP, Staddon AP, Baker LH, Schuetze SM, Tolcher AW, D’Amato GZ, et al. Estudio de fase II de la diana en mamíferos del inhibidor de la rapamicina ridaforolimus en pacientes con sarcomas óseos y de tejidos blandos avanzados. J Clin Oncol (2012) 30(1):78–84. doi:10.1200/JCO.2011.35.6329
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
85. Grignani G, Palmerini E, Dileo P, Asaftei SD, D’Ambrosio L, Pignochino Y, et al. Un ensayo de fase II de sorafenib en osteosarcoma de alto grado recidivante e irresecable después del fracaso de la terapia multimodal estándar: un estudio del Grupo Italiano de Sarcoma. Ann Oncol (2012) 23(2):508–16. doi:10.1093/annonc/mdr151
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
86. Malempati S, Weigel B, Ingle AM, Ahern CH, Carroll JM, Roberts CT, et al. Ensayo de fase I/II y estudio farmacocinético de cixutumumab en pacientes pediátricos con tumores sólidos refractarios y sarcoma de Ewing: un informe del Children’s Oncology Group. J Clin Oncol (2012) 30(3):256–62. doi:10.1200/JCO.2011.37.4355
Resumen de Pubmed | Pubmed Texto Completo | Texto completo de CrossRef
Cita: Guijarro MV, Ghivizzani SC y Gibbs CP (2014) Modelos animales en osteosarcoma. Frente. Oncol.4:189. doi: 10.3389/fonc.2014.00189
Recibido: 31 Mayo 2014; Aprobado: 07 Julio 2014;
Publicado en línea: 18 de julio de 2014.
Editado por:
Amancio Carnero, Instituto de Biomedicina de Sevilla, España
Revisado por:
Carmen Blanco Aparicio, Centro Nacional de Investigaciones Oncológicas, España Irene Ferrer, IBIS, España
Copyright: © 2014 Guijarro, Ghivizzani y Gibbs. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia de Atribución Creative Commons (CC BY).
*Correspondencia: Maria V. Guijarro, Departamento de Ortopedia y Rehabilitación, Universidad de Florida, 1600 Archer Road, MSB M2-212, Gainesville, FL 32610, EE.UU. correo electrónico: guijam@ufl.edu
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o reclamo que pueda ser hecho por su fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía