
Perfil metagenómico de las comunidades virales y microbianas de las lesiones de la viruela ovina
 Perfil metagenómico de las comunidades virales y microbianas de las lesiones de la viruela del virus de la enfermedad cutánea contagiosa y de los huéspedes infectados por el virus de la viruela ovina
Perfil metagenómico de las comunidades virales y microbianas de las lesiones de la viruela del virus de la enfermedad cutánea contagiosa y de los huéspedes infectados por el virus de la viruela ovina
 Fedor S. Sharko1
Fedor S. Sharko1  Ali Mazloum2
Ali Mazloum2  Alena O. Krotova2 Olga P. Byadovskaya2 Larisa B. Prokhvatilova2
Alena O. Krotova2 Olga P. Byadovskaya2 Larisa B. Prokhvatilova2  Ilya A. Chvala2
Ilya A. Chvala2  Ustin E. Zolotikov1 Alexandra D. Kozlova1
Ustin E. Zolotikov1 Alexandra D. Kozlova1  Anastasia S. Krylova1
Anastasia S. Krylova1  Erika V. Grosfeld1,3 Anastasia V. Prokopenko1
Erika V. Grosfeld1,3 Anastasia V. Prokopenko1  Aleksei A. Korzhenkov1 Maxim V. Patrushev1
Aleksei A. Korzhenkov1 Maxim V. Patrushev1  Zorigto B. Namsaraev1
Zorigto B. Namsaraev1  Alexander V. Sprygin2
Alexander V. Sprygin2  Stepan V. Toshchakov1*
Stepan V. Toshchakov1*- 1Centro Nacional de Investigación «Instituto Kurchatov», Moscú, Rusia
- 2Centro Federal de Sanidad Animal FGBI ARRIAH, Vladimir, Rusia
- 3Instituto de Física y Tecnología de Moscú, Universidad Nacional de Investigación, Dolgoprudny, Rusia
Introducción: Se ha reconocido que las infecciones por capripoxvirus tienen un fuerte tropismo cutáneo con la manifestación de lesiones cutáneas en forma de nódulos y costras en los respectivos huéspedes, seguidas de necrosis y descamación. Teniendo en cuenta que la microbiota cutánea es una comunidad compleja de bacterias, hongos y virus comensales que se ven influenciados por infecciones que conducen a estados patológicos, no hay pruebas sobre cómo se ve afectado el microbioma cutáneo durante la patogénesis del capripoxvirus.
Métodos: En este estudio, se utilizó la secuenciación metagenómica de escopeta para investigar el microbioma en las lesiones de viruela de huéspedes infectados con el virus de la enfermedad de la piel nodular contagiosa y el virus de la viruela ovina.
Resultados: El análisis reveló un alto grado de variabilidad en las estructuras de la comunidad bacteriana en las muestras de piel afectadas, lo que indica la importancia de que los microorganismos comensales específicos colonicen a los huéspedes individuales. Las bacterias más comunes y abundantes encontradas en las muestras de sarna fueron Fusobacterium necrophorum, Streptococcus dysgalactiae, Helcococcus ovis y Trueperella pyogenes, independientemente del hospedador. Se identificaron lecturas bacterianas pertenecientes a los géneros Moraxella, Mannheimia, Corynebacterium, Staphylococcus y Micrococcus.
Discusión: Este estudio es el primero en investigar los cambios asociados al virus capripox en el microbioma de la piel utilizando perfiles metagenómicos del genoma completo. Los hallazgos proporcionarán una base para futuras investigaciones sobre la patogénesis del capripoxvirus. Además, este estudio pone de manifiesto el reto de seleccionar un enfoque bioinformático óptimo para el análisis de datos metagenómicos en la práctica clínica y veterinaria. Por ejemplo, la clasificación directa de lecturas mediante un algoritmo basado en kmer dio lugar a un número significativo de falsos positivos sistemáticos, que pueden atribuirse a las peculiaridades del algoritmo y a la selección de la base de datos. Por el contrario, el proceso de ensamblaje de novo requiere un gran número de lecturas objetivo de la comunidad microbiana simbiótica. En este trabajo, los datos de secuenciación obtenidos se procesaron mediante tres enfoques diferentes, incluida la clasificación directa de lecturas basada en k-mers, el mapeo de lecturas a una base de datos de genes marcadores y el ensamblaje y agrupamiento de novo de contigs metagenómicos. Las ventajas y desventajas de estas técnicas y su practicidad en el ámbito veterinario se discuten en relación con los resultados obtenidos.
1 Introducción
Los virus capripox son infecciones virales altamente contagiosas y a menudo mortales de ovejas y vacas. Deben notificarse a la Organización Mundial de Sanidad Animal (OMSA) (1). Estos virus pertenecen al género Poxviridae Capripoхvirus, que incluye el virus de la viruela ovina (SPV), el virus de la viruela caprina (GTPV) y el virus de la enfermedad cutánea nodular contagiosa (LSDV) (2, 3). Las infecciones relacionadas con el Capripoxvirus, que se descubrieron por primera vez en África, se descubrieron por primera vez en África y desde entonces se han extendido rápidamente a Europa y Asia (4). Los genomas de los virus Capripox consisten en ADN bicatenario de aproximadamente 150 pares de kilobases de longitud, con repeticiones terminales en cada extremo, que codifican 147 marcos de lectura abiertos (ORF) (3). Los genomas de las tres especies están estrechamente relacionados, mostrando un 97% de identidad nucleotídica. El alto nivel de similitud entre los poxvirus aumenta la probabilidad de recombinación en el campo (5, 6). Sin embargo, los capripoxvirus exhiben diferentes niveles de adaptación del huésped. Si bien se han observado casos de LSD en antílopes y jirafas, sus huéspedes principales son el ganado y los búfalos de agua (7). A su vez, se ha descubierto que el GTPV y el SPPV poseen capacidad de infección cruzada natural o experimental, causando enfermedades en ambas especies hospedadoras, cabras y ovejas (8).
Las ovejas y cabras afectadas desarrollan fiebre y diversos grados de generalización. Los párpados se hinchan y la secreción mucopurulenta forma costras en las fosas nasales. Se desarrollan lesiones cutáneas generalizadas que se ven fácilmente en el hocico, las orejas y las áreas libres de lana (9). Las lesiones comienzan como áreas eritematosas en la piel libre de lana y progresan rápidamente a placas circulares elevadas con bordes congestionados (9). Tras la necropsia, las observaciones a menudo mostraron edema y congestión pulmonar, así como nódulos presentes en los pulmones y debajo de la piel en los músculos (10). A medida que las lesiones comienzan a retroceder, se produce necrosis, seguida de la formación de costras que son abundantes en partículas virales (11). En condiciones experimentales, hasta el 50% de los animales infectados por el virus del capripox no desarrollan los nódulos típicos y permanecen no infectados o infectados subclínicamente (12).
A pesar de la creciente cantidad de investigaciones sobre los capripoxvirus, todavía existen brechas considerables en el conocimiento, especialmente en lo que respecta a la diversidad genética, la especificidad del huésped, las vías de transmisión y las diferencias en la gravedad de los síntomas entre los brotes de campo y los entornos experimentales (12-15). Por lo general, la piel alberga diferentes comunidades bacterianas y virales que pueden servir como sujetos para la investigación metagenómica tanto en condiciones naturales como experimentales (16). Sin embargo, a pesar de que las lesiones cutáneas o costras son el rasgo característico de las enfermedades por capripoxvirus, existe una falta de conocimiento publicado sobre las comunidades bacterianas y virales presentes en las lesiones inducidas por capripox. La investigación de posibles coinfecciones y la identificación de nuevos agentes durante la infección por capripoxvirus podrían proporcionar un valioso conocimiento de los mecanismos de patogénesis natural subyacentes. Es importante considerar la probabilidad de infecciones bacterianas y virales secundarias, que comúnmente surgen como factores de complicación para los capripoxvirus, por lo que requieren tratamiento antibiótico (17).
Los enfoques metagenómicos se utilizan progresivamente en la práctica clínica (18, 19) y veterinaria para el seguimiento de las coinfecciones y las infecciones oportunistas. Además de la monitorización directa de la coinfección, los métodos metagenómicos se han utilizado con éxito para analizar la especificidad del huésped y la diversidad genética de los agentes etiológicos nuevos y caracterizados. La revisión realizada por Suminda y sus coautores en 2022 proporciona más información sobre este tema (20). Otra aplicación del enfoque metagenómico es el análisis de las vías de transmisión de infecciones zoonóticas, que puede realizarse incluso utilizando muestras ambientales tomadas fuera del huésped (21). El método más rentable y adecuado para el seguimiento de las coinfecciones y el análisis generalizado de la composición de la comunidad microbiana es la secuenciación altamente paralela de genes marcadores (por ejemplo, ARNr 16S). Sin embargo, este método tiene sus limitaciones, incluida la baja resolución taxonómica y la capacidad de evaluar solo un grupo específico de patógenos, lo que lleva a una interpretación restringida de los hallazgos (22). La secuenciación metagenómica de escopeta permite el análisis simultáneo de bacterias, virus y microorganismos eucariotas en un conjunto de datos, lo que proporciona un análisis filogenético de alta precisión (23). El principal reto de esta técnica es la elección de un algoritmo fiable y eficiente en cuanto a datos para el análisis de la comunidad microbiana (23). Por un lado, la clasificación de lectura directa requiere datos mínimos y permite identificar la composición de la comunidad muestral; Sin embargo, por otro lado, este enfoque con frecuencia arroja resultados falsos positivos (24). Por otro lado, el ensamblaje de novo de los genomas microbianos permite una determinación muy precisa de la filogenia y el análisis de la posible resistencia a los antibióticos (25). Sin embargo, este enfoque requiere una gran cantidad de datos y múltiples muestras para la agrupación de cobertura diferencial de los contigs metagenómicos.
Como se ha indicado anteriormente, el objetivo de este estudio es analizar el microbioma presente en las costras cutáneas inducidas por SPV/LSDV mediante secuenciación metagenómica de escopeta. En este estudio se utilizaron tres enfoques independientes para analizar los datos metagenómicos obtenidos mediante la secuenciación de muestras clínicas, a saber, la clasificación de lecturas a través de k-mers, la clasificación de lecturas a través del mapeo a una base de datos de genes marcadores, y el ensamblaje y agrupamiento de novo de contigs metagenómicos. A partir de los hallazgos obtenidos, se discuten las ventajas e inconvenientes de estos métodos bioinformáticos para aplicaciones clínicas.
2 Materiales y métodos
2.1 Descripción de las muestras
Se tomaron doce muestras de costra cutánea de animales que mostraban síntomas de infección por SPPV de diferentes regiones de la Federación de Rusia y de diferentes huéspedes (Bos taurus y Ovis aries). Las muestras se enviaron al laboratorio de referencia del Instituto Federal de Sanidad Animal (FGBI «ARRIAH») para la confirmación molecular de la infección por SPPV o LSDV. Los criterios de selección de las muestras fueron los siguientes: debían ser entregadas a FGBI ARRIAH en hielo frío y en cantidad suficiente (al menos 200 mg de biomasa). La cadena de frío es crucial para preservar la comunidad microbiana inherente a un animal vivo, sin ningún artefacto causado por el crecimiento excesivo de la microflora secundaria. Dado que las muestras se obtienen con frecuencia de regiones remotas con una logística deficiente, rara vez se respeta la cadena de frío. Por lo tanto, solo pudimos recolectar tres muestras de ovejas y nueve bovinos para este estudio. Las 12 muestras dieron positivo para el genoma del SPPV utilizando un kit de PCR interno (26) (Tabla 1).
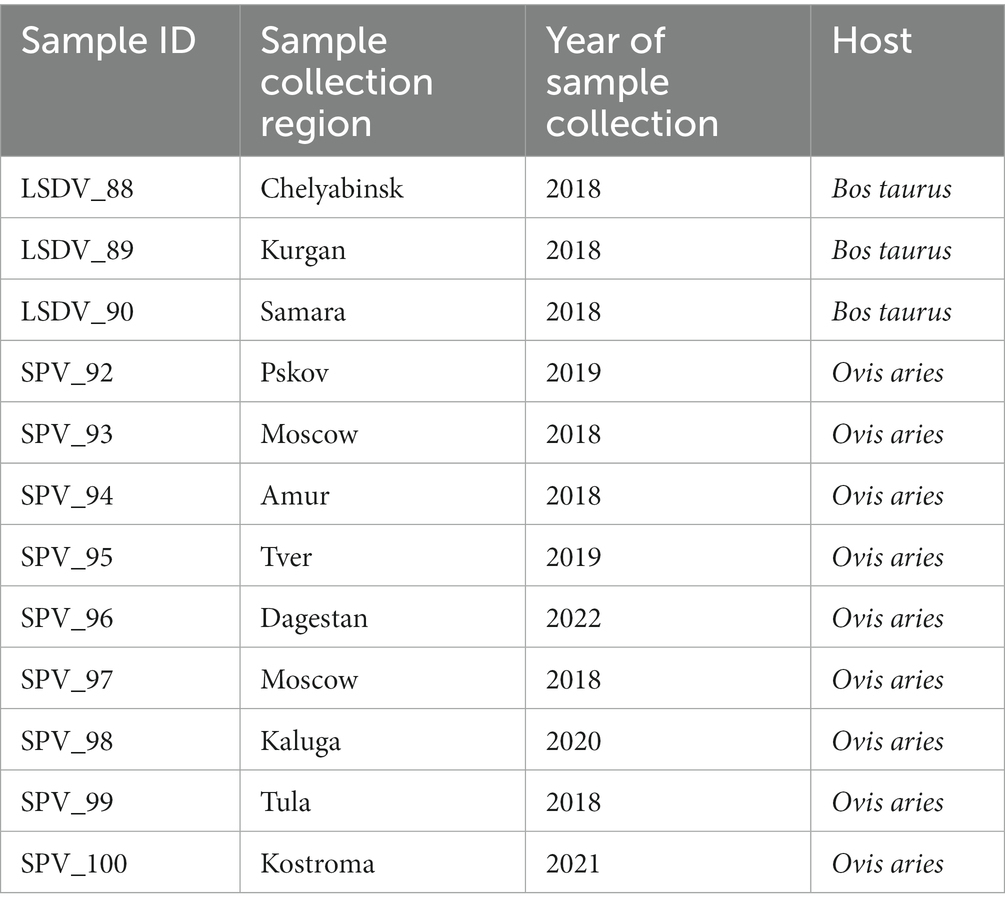 Tabla 1. Características de la muestra.
Tabla 1. Características de la muestra.
2.2 Extracción de ADN y preparación de la biblioteca de fragmentos
El ADN metagenómico se extrajo con el kit de microbioma de ADN QIAamp (Qiagen, Alemania), de acuerdo con las instrucciones del fabricante. Se eligió el kit especificado porque permite el enriquecimiento de la fracción microbiana de la muestra mediante lisis diferencial de células eucariotas. La cantidad y calidad del ADN se evaluó utilizando NanoDrop™ Espectrofotómetro 8.000 (Thermo Fisher Scientific, Carlsbad, CA, Estados Unidos). La relación A260/280 estaba en el rango de 1,8-1,9, mientras que la A260/230 estaba en el rango de 1,8-2,2, lo que refleja una pureza suficiente de las preparaciones de ADN. Se utilizaron 400 nanogramos de ADN de cada muestra como insumo para la preparación de la biblioteca. El ADN se fragmentó en fragmentos de 250-320 pb con el ultrasonido Focalizado Covaris ME220 (Covaris, Woburn, MA, Estados Unidos) de acuerdo con el protocolo de sonicación, proporcionado por el fabricante. Se seleccionó el método de fragmentación ultrasónica porque minimiza los sesgos en la composición de la comunidad microbiana que pueden surgir del rendimiento subóptimo de la fragmentasa para genomas ricos en AT o GC. Las bibliotecas compatibles con DNBSeq de fragmentos estándar se prepararon utilizando el kit de preparación de bibliotecas de ADN universal MGIEasy y luego se circularizaron con el kit de circularización MGIEasy (ambos, MGITech, República Popular China), de acuerdo con las instrucciones proporcionadas por el fabricante.
2.3 Secuenciación y control de calidad
La secuenciación se realizó mediante la plataforma MGITECH DNBSEQ 400 (MGITECH, República Popular China), que mostró excelentes resultados en estudios metagenómicos de referencia (27). Para la secuenciación se utilizó el kit de reactivos DNBSEQ-G400RS High-throughput Sequencing Set (PE150), ya que era la longitud de lectura máxima disponible en el momento de los experimentos. La demultiplexación de las muestras indexadas se realizó en el instrumento. El control de calidad se realizó mediante el análisis de los datos brutos de calidad de lectura con las herramientas seqkit (28) y FastQC (29).
2.4 Elaboración de perfiles comunitarios mediante la clasificación directa de lecturas metagenómicas
Las lecturas metagenómicas obtenidas se recortaron y filtraron por calidad con fastp versión 0.23.4 (30) utilizando los siguientes parámetros: calidad media mimal en la ventana deslizante de 4 pb frontal y trasera: 25; número máximo de bases no identificadas en la lectura: 1; calidad media mínima en lectura recortada: 20; Longitud mínima de lectura recortada: 50. Las lecturas del huésped y las lecturas correspondientes a la contaminación del ADN humano se detectaron mediante el mapeo de lecturas al genoma del huésped o de la contaminación (Homo sapiens, Bos taurus y Ovis aries) utilizando Bowtie2 (31).
Antes de utilizar el clasificador Kraken2, se creó una base de datos personalizada, que incluía arqueas, bacterias, virus, protozoos, genomas completos de plásmidos de hongos, UniVec Core y genomas de mamíferos, incluidos genomas humanos, vacas y ovejas. Esta base de datos se utilizó para que el Kraken2 clasificara todas las lecturas, proporcionando a cada lectura su identificación taxonómica. Para obtener resultados de clasificación fiables utilizando el Kraken2, se utilizaron parámetros de confianza = 0,3 y grupos de aciertos mínimos = 3. Para estimar la abundancia de especies en cada muestra se utilizó helecho con los parámetros: -r 100 -t 50 (32). Utilizando el script KrakenTools alpha_diversity.py, se calcularon las α diversidades de Berger-Parker, Fisher, Simpson, Simpson inversa y Shannon para cada muestra (33).
Para la clasificación de las lecturas por genes marcadores específicos del clado con Metaphlan4 (34), se clasificaron pares de lecturas de secuenciación temporizada y filtrada con respecto a la última versión de la base de datos Metaphlan4 con parámetros de clasificación estándar.
2.5 Ensamblaje de novo, agrupamiento metagenómico y análisis filogenético
Cada muestra secuenciada se ensambló por separado. Las lecturas recortadas y filtradas se ensamblaron con el ensamblador metaSPAdes de novo (35) utilizando tamaños k-mer de 99 y 127. La elección de una mayor longitud de kmer en comparación con los valores predeterminados implementados en SPAdes para el ensamblaje metagenómico se debió a que la longitud de lectura fue de 150 nucleótidos, lo que permitió utilizar valores más altos para obtener un mejor ensamblaje (36). El binning de los contigs metagenómicos se realizó mediante los algoritmos Concoct (37), Metabat2 (38) y Maxbin 2.0 (39), implementados en el pipeline Metawrap (40). Se realizó una agrupación adicional con la herramienta de agrupación pyYamb desarrollada internamente (41). El refinamiento de los conjuntos de bins obtenidos se realizó con el paquete de herramientas DAS (42). La taxonomía de los bins resultantes se determinó con GTDB-toolkit (43). La completitud y el nivel de contaminación de los contenedores resultantes se estimó con el paquete CheckM2 (44).
El árbol filogenético de 41 genes marcadores se reconstruyó de la siguiente manera: Los modelos de Markov de genes marcadores se adquirieron de la base de datos Pfam (45). Los genes se buscaron mediante secuencias de aminoácidos utilizando hmmscan v.3.3.2 con un valor e de 1e-10 (46). Las secuencias de nucleótidos correspondientes para cada marcador se alinearon automáticamente utilizando MAFFT v.7.520 (47). El árbol filogenético se desarrolló utilizando el modelo evolutivo GTR con Fasttree v.2.1.11 (48) con 1.000 bootstraps.
La extracción de las secuencias utilizadas para la MLST de S. dysgalactiae se realizó con el paquete FastMLST (49). El alineamiento y la reconstrucción de la filogenia se realizaron con mafft (47) e IQTree con 1.000 botstraps (50), respectivamente. Todos los árboles filogenéticos se visualizaron con el servidor web iTOL (51). El análisis de ANI y la visualización de mapas de calor se realizaron con el paquete pyANI (52).
El análisis de los genes de resistencia a antibióticos en los bins metagenómicos obtenidos se realizó con el paquete staramr (53).
3 Resultados
3.1 Análisis de la diversidad de costras cutáneas mediante clasificación metagenómica directa de lecturas
3.1.1 Secuenciación metagenómica y contaminación del huésped
El estudio analizó el microbioma de las costras cutáneas de la dermatosis nodular contagiosa y la viruela ovina utilizando muestras enviadas a FGBI ARRIAH de varias regiones de Rusia para la confirmación por PCR de infecciones por capripoxvirus. Se aplicaron estrictos criterios de selección a la condición de los especímenes de biomasa en el momento de su recepción, debido a la posibilidad de sobrecrecimiento de microflora secundaria en muestras tomadas en condiciones de granja no estériles (ver Métodos). Debido a este hecho, solo se seleccionaron tres muestras de biomasa de sarna bovina y nueve muestras de sarna ovina (Tabla 1).
La secuenciación de bibliotecas metagenómicas de escopeta con el secuenciador DNBSeq-400 dio como resultado entre 10,2 y 34,3 millones de pares de lectura por muestra. La eliminación de las lecturas del huésped reveló altos niveles de contaminación con ADN del huésped que oscilaban entre el 57,0 y el 96,7% (Tabla 2). El filtrado adicional de lecturas relacionadas con humanos, recomendado para animales de granja y domésticos (54), mostró la contaminación con ADN humano en el 0,15-2,56% (0,64% en promedio). Tras el filtrado de calidad y la eliminación de la contaminación relacionada con el huésped y los seres humanos, se dispuso de entre 0,31 y 9,15 millones de pares de lectura para la clasificación directa y el ensamblaje de novo.
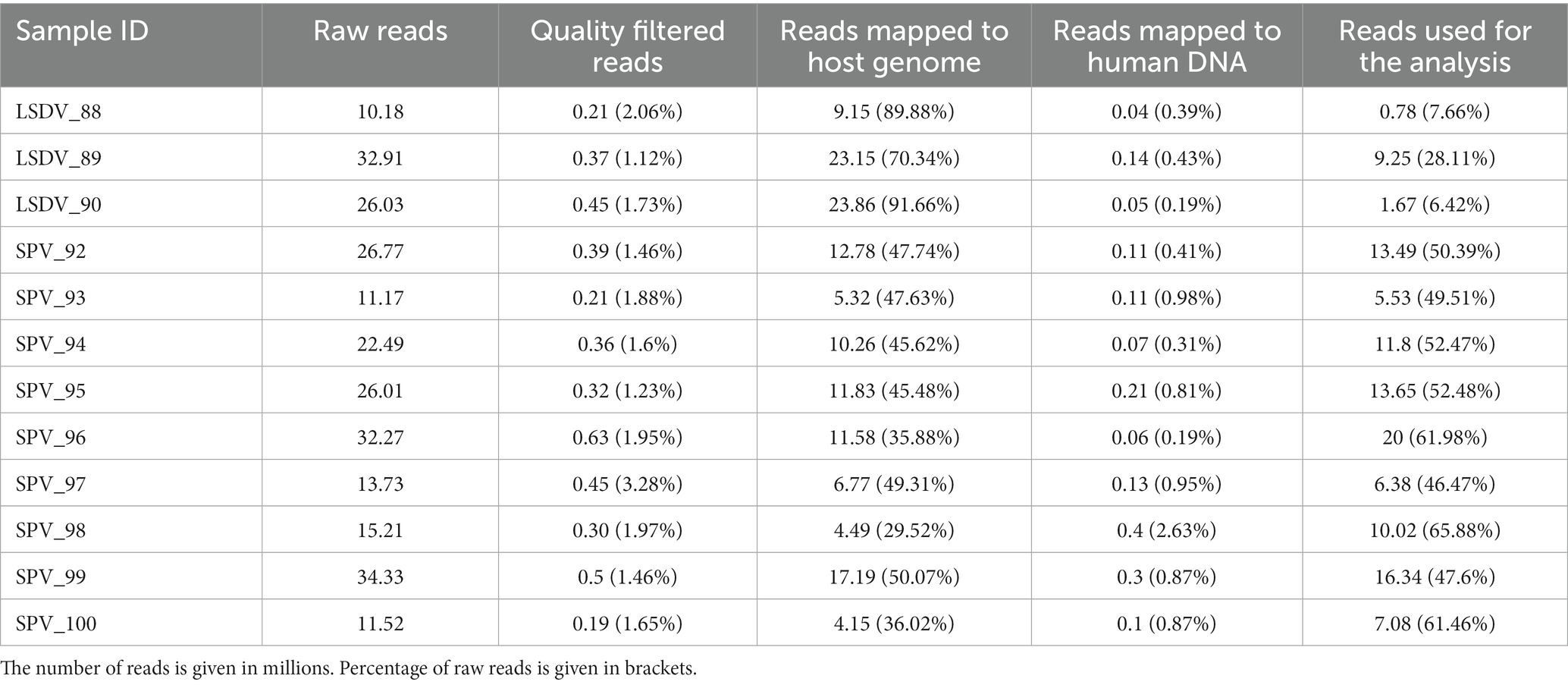 Tabla 2. Procesamiento de lecturas metagenómicas.
Tabla 2. Procesamiento de lecturas metagenómicas.
3.1.2 Diversidad alfa
La clasificación metagenómica directa de las lecturas, realizada con Kraken2 (55) utilizando la base de datos RefSeq personalizada complementada con genomas del huésped (ver Métodos), dio como resultado la clasificación del 14,7-67,2% de las lecturas filtradas (Tabla suplementaria 1). Sin embargo, a pesar del filtrado previo de las lecturas mediante el mapeo del genoma del huésped, entre el 5,35 y el 28,71 % de las lecturas filtradas se clasificaron como relacionadas con el huésped. Cabe señalar que entre el 5,35 y el 28,71 % de las lecturas filtradas se clasificaron como relacionadas con el host. Para verificar los hallazgos de Kraken2 mediante un algoritmo de clasificación diferente, los datos se verificaron utilizando el paquete Metaphlan4. Este paquete implementa un algoritmo que se basa en el mapeo de lectura a genes marcadores de copia única (34). Los genes marcadores representan una pequeña fracción del genoma, lo que resulta en un menor porcentaje de lecturas clasificadas para Metaphlan4 en comparación con Kraken2. En consecuencia, Metaphlan4 detecta mal los microorganismos infrarrepresentados, lo que conduce a métricas de diversidad alfa más bajas que Kraken2. Al mismo tiempo, las métricas de diversidad alfa para muestras con más de 10 millones de lecturas objetivo fueron comparables entre Kraken2 y Metaphlan4 (Figura 1; Cuadro complementario 2). Varios estudios de referencia han encontrado que Metaphlan4 exhibe una mayor especificidad pero una menor sensibilidad que Kraken2 (56). En consecuencia, las métricas de diversidad alfa basadas en Metaphlan4 fueron significativamente más bajas que las derivadas de los resultados de Kraken2, con un valor medio del índice de Shannon de 1,38 para Metaphlan4 y de 2,06 para Kraken2. El número promedio de especies detectadas también fue mucho menor, con 16 para Metaphlan4 y 61 para Kraken2 (Figura 1). Las métricas de diversidad alfa no mostraron diferencias significativas entre los animales infectados por LSDV y los animales infectados por SPV (Figura 1). Desafortunadamente, debido a las características específicas del procedimiento de recogida de muestras, no fue posible comparar el nivel de diversidad de la microbiota para las zonas de piel sanas y afectadas de cada animal. Varios estudios sobre el microbioma de las lesiones cutáneas en humanos indican una disminución de la diversidad alfa en las zonas afectadas debido a la prevalencia de microorganismos oportunistas (57, 58). Aunque hay poca investigación sobre las enfermedades de la piel en animales de granja, hay evidencia de una disminución de la diversidad en las lesiones cutáneas causadas por la dermatitis por hendidura de la ubre en vacas lecheras (59). Esto podría explicar en parte las bajas métricas de diversidad alfa observadas en este estudio.
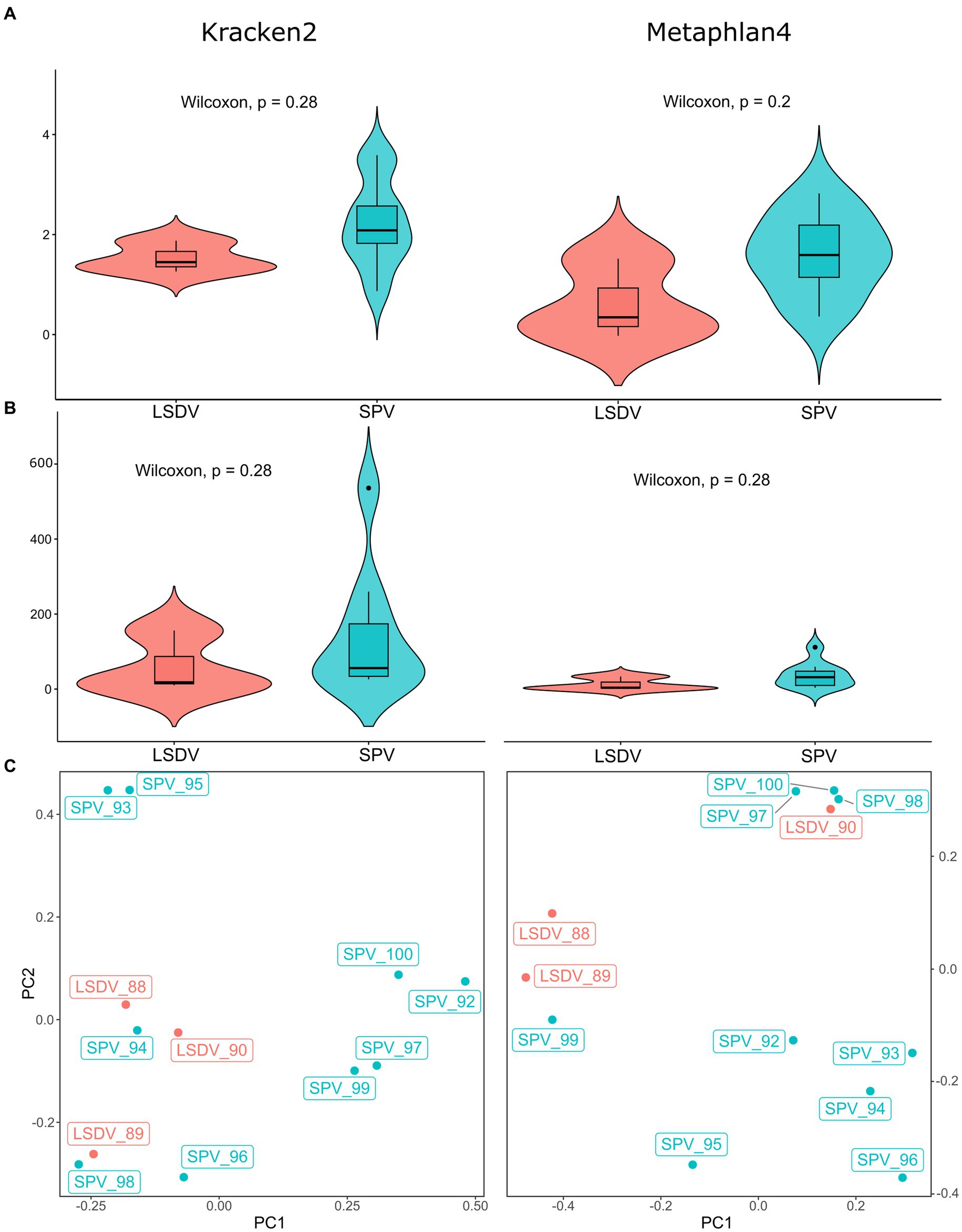 Figura 1. Diversidad alfa y beta de las comunidades microbianas de las costras de la piel de animales infectados con LSDV y SPV. (A) Diagramas de violín de los índices de Shannon comparados entre animales infectados con LSDV y SPV. Los índices basados en los resultados de Kraken2 se muestran en el panel izquierdo, los índices basados en Metaphlan4 se muestran en el panel derecho. (B) Gráficos de violín del número de especies comparadas entre animales infectados por LSDV y SPV. Los valores basados en los resultados de Kraken2 se muestran en el panel izquierdo, los valores basados en Metaphlan4 se muestran en el panel derecho. (C) Análisis de componentes principales de las comunidades microbianas de costras cutáneas.
Figura 1. Diversidad alfa y beta de las comunidades microbianas de las costras de la piel de animales infectados con LSDV y SPV. (A) Diagramas de violín de los índices de Shannon comparados entre animales infectados con LSDV y SPV. Los índices basados en los resultados de Kraken2 se muestran en el panel izquierdo, los índices basados en Metaphlan4 se muestran en el panel derecho. (B) Gráficos de violín del número de especies comparadas entre animales infectados por LSDV y SPV. Los valores basados en los resultados de Kraken2 se muestran en el panel izquierdo, los valores basados en Metaphlan4 se muestran en el panel derecho. (C) Análisis de componentes principales de las comunidades microbianas de costras cutáneas.
3.1.3 Diversidad beta
El análisis de PERMANOVA de la diversidad beta utilizando la función adonis2 del paquete vegano no reveló diferencias significativas entre los animales infectados con LSDV y SPV, ni para Kraken2 (R2 = 0,114, valor de p > 0,1, con 999 permutaciones) ni para Metaphlan4 (R2 = 0,126, valor de p > 0,01, con 999 permutaciones). El análisis de componentes principales de los resultados tampoco mostró una agrupación de muestras clara (Figura 1; Cuadro complementario 4). Este resultado indica que la estructura de la comunidad microbiana presente en las costras está determinada principalmente por el azar, influenciado por la prevalencia de un patógeno oportunista bacteriano específico dentro del área de la lesión. Sin embargo, no se ve afectado por el tipo específico de capripoxvirus presente.
3.2 Composición del microbioma de las costras de la piel
3.2.1 Microbioma central
Las lecturas de secuenciación clasificadas por Kraken2 fueron claramente predominadas por las correspondientes al agente infeccioso principal (Figura 2). En particular, se observó la clasificación de un pequeño número de lecturas a SPV en animales infectados con LSDV y viceversa. Sin embargo, el análisis de ensamblaje de novo no confirmó la presencia de los dos virus, lo que nos permite atribuir este resultado a falsos positivos causados por la baja especificidad del algoritmo basado en kmer implementado en Kraken2. Curiosamente, se descubrió un caso de coinfección viral de ovejas (muestra SPV_99) con el virus orf (60) (Figura 2).
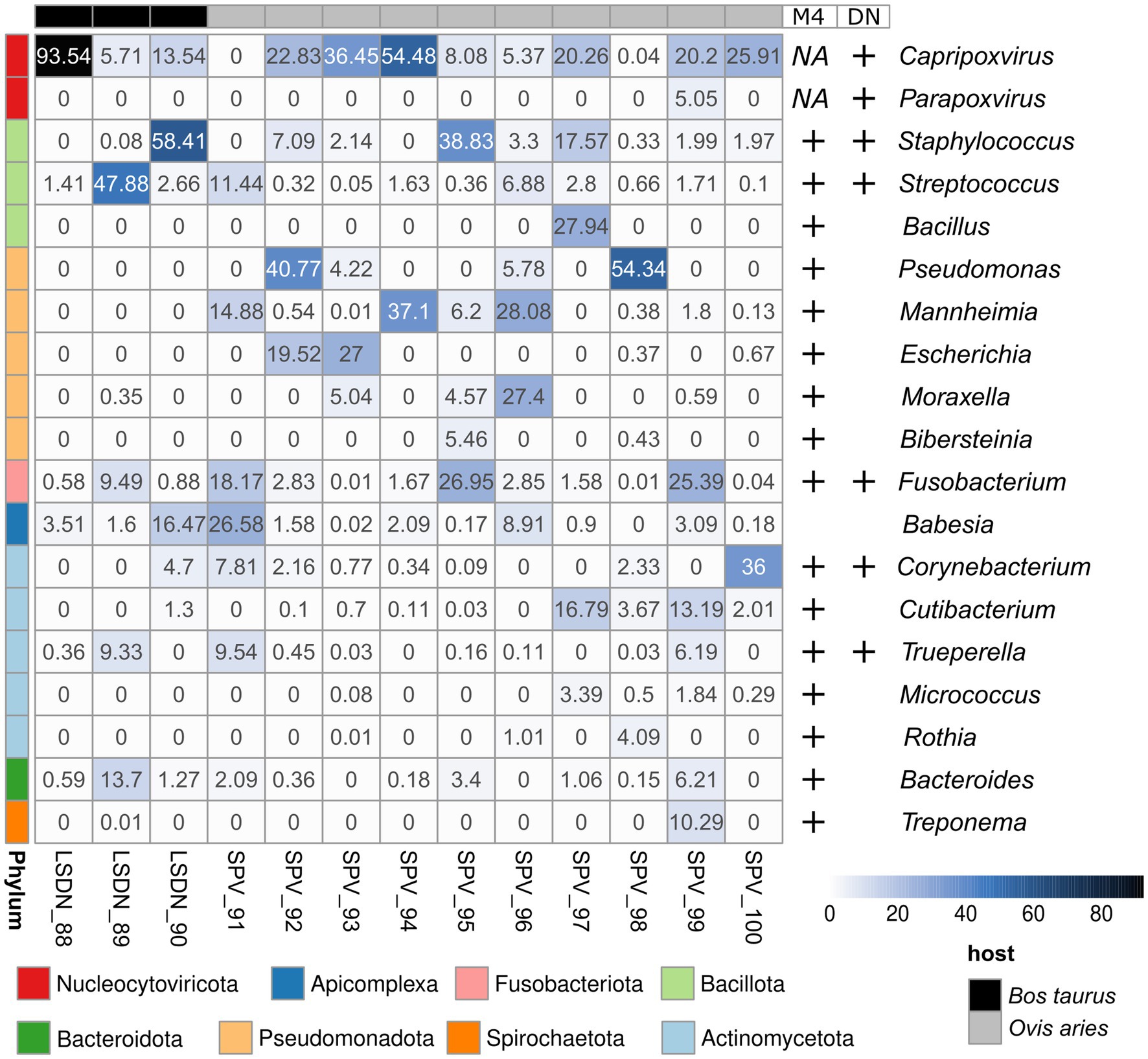 Figura 2. Mapa de calor de abundancia de lecturas clasificadas de Kraken2. Los valores de abundancia se muestran en porcentajes. Los resultados, respaldados por Metaphlan4 (M4) o el ensamblaje metagenómico de novo y la clasificación de bin (DN), se resaltan con signos ‘+’ en el lado derecho del mapa de calor.
Figura 2. Mapa de calor de abundancia de lecturas clasificadas de Kraken2. Los valores de abundancia se muestran en porcentajes. Los resultados, respaldados por Metaphlan4 (M4) o el ensamblaje metagenómico de novo y la clasificación de bin (DN), se resaltan con signos ‘+’ en el lado derecho del mapa de calor.
El único microorganismo eucariota identificado fue Babesia bigemina, detectable en todas las muestras analizadas. El número de lecturas asignadas osciló entre el 0,003 y el 16,5% de todas las lecturas clasificadas de Kraken2 (Figura 2; Figura complementaria 1). Sin embargo, los resultados de Metaphlan4 no respaldaron este hallazgo (Figura suplementaria 2; Tabla 3). Un análisis más detallado de las lecturas asignadas a Babesia bigemina por Kraken2 mostró que todas estaban asignadas al pequeño andamio del genoma de B. bigemina, pero no a los cromosomas (Tabla suplementaria 5). Este hecho podría explicarse por la contaminación procariota del ensamblaje de B. bigemina, depositado en NCBI RefSeq e importado en la base de datos Kraken2.
Los microorganismos procariotas dominantes variaron de una muestra a otra, según Kraken2/Metaphlan4 (Figura 2; Cuadro complementario 3; Figuras 1 y 2). El análisis de los miembros principales de las comunidades mostró que la única especie, detectada por todas las herramientas en más del 50% de las muestras con una abundancia superior al 1%, fue Fusobacterium necrophorum, que se reportó como causante de la enfermedad de pudrición del pie en bovinos (61) y ovinos (62) (Tabla 3). Otros taxones principales variaron según el grupo de muestra (SPV/LSDV) y el método de análisis (Kraken2/Metaphlan4). En el caso de los animales infectados con LSDV, Kraken2 detectó Streptococcus dysgalactiae, que se sabe que causa mastitis en el ganado bovino (63), en todas las muestras, pero solo en las muestras de animales infectados con LSDV prevalecieron los microorganismos (Figura 2). A su vez, 7 de las 9 muestras de ovejas demostraron la presencia del patógeno oportunista común Staphylococcus aureus capaz de causar el desarrollo de mastitis y otras enfermedades de la piel en el ganado (64, 65). Bacteroides heparinolyticus, reportado como asociado con metritis de vacas lecheras (66) fue detectado en 9 de las 12 muestras analizadas. Las ovejas infectadas por SPV mostraron un aumento significativo de la abundancia de Cutibacterium, en comparación con las vacas (Figura 2). A su vez, los resultados de Metaphlan4 mostraron la presencia de Helcococcus ovis en 2 de 3 muestras de costra de LSDV y 3 de 9 muestras de SPV. Curiosamente, según Metaphlan4, H. ovis dominó absolutamente la comunidad microbiana en la LSDV_88 muestral, alcanzando el 100% de las lecturas procariotas clasificadas (Figura suplementaria 2). Recientemente se ha informado de que las bacterias, aisladas por primera vez en el pulmón y el hígado de ovejas post mortem (67), están asociadas con enfermedades pulmonares de las ovejas (68) y causan abortos en el ganado vacuno (69). La detección de H. ovis fue apoyada por la clasificación taxonómica basada en el genoma de bins metagenómicos ensamblados de novo, por lo que suponemos que el resultado es falso negativo de Kraken2. Por lo tanto, todas las bacterias identificadas como miembros de la comunidad central de lesiones cutáneas asociadas al capripoxvirus, fueron previamente reportadas como asociadas con enfermedades de la piel del ganado.
 Tabla 3. Miembros centrales de la microbiota de cicatrices asociadas al capripoxvirus.
Tabla 3. Miembros centrales de la microbiota de cicatrices asociadas al capripoxvirus.
3.2.2 Microbioma específico de la muestra
Los miembros de la comunidad microbiana de la sarna detectados esporádicamente en diferentes muestras eran muy diversos. A nivel de género, se detectaron 172 géneros utilizando Kraken 2. Al mismo tiempo, un análisis de genes marcadores más específicos utilizando Metaphlan4 permitió la detección de solo 54 géneros válidos. El ensamblaje de novo y el posterior binning de contigs metagenómicos permitieron obtener 10 canastas metagenómicas de calidad aceptable. Así, se detectaron 44 géneros microbianos mediante al menos dos de los tres enfoques utilizados (Figura 3).
 Figura 3. Diagrama de Venn proporcional al área de los géneros detectados por diferentes enfoques bioinformáticos. El diagrama se dibujó con la aplicación web DeepVenn (70).
Figura 3. Diagrama de Venn proporcional al área de los géneros detectados por diferentes enfoques bioinformáticos. El diagrama se dibujó con la aplicación web DeepVenn (70).
Los miembros mencionados anteriormente de la parte central de la comunidad microbiana, a saber, Fusobacterium, Streptococcus, Staphylococcus, Bacteroides y Helcococcus, fueron detectados por los tres métodos de análisis. La abundancia y prevalencia de estas bacterias fueron comparables para todos los enfoques, excepto para Helcococcus, cuya abundancia supuestamente no fue estimada por Kraken2 debido a la ausencia del genoma de Helcococcus ovis en la base de datos de referencia (algunas lecturas fueron clasificadas por Kraken2 como Helcococcus bovis). Otras bacterias, detectadas tanto por la clasificación directa de las lecturas de secuenciación como por el ensamblaje y agrupamiento de novo, pertenecían a los géneros Trueperella (hasta el 9,8% de las lecturas clasificadas por Kraken2), Corynebacterium (hasta el 56% de las lecturas clasificadas por Kraken2), Oligella (hasta el 3,7%) y Porphyromonas (hasta el 1,1%).
Los géneros microbianos identificados por ambos enfoques de clasificación de lectura directa, a saber, Kraken2 y Metaphlan4, pero no por agrupamiento metagenómico, también pueden caracterizarse por una alta abundancia media o por el hecho de que la mayoría de ellos estaban presentes en al menos un tercio de las muestras analizadas. Los géneros Pseudomonas y Mahnheimia fueron los más abundantes, la proporción de lecturas asignadas a estos géneros alcanzó el 76 y 82%, respectivamente y se detectaron en al menos un tercio de las muestras (Figura 3). Los representantes de Cutibacterium, a nivel de especie determinado principalmente como С. acnes, alcanzaron el 12,9 y el 21,1% de abundancia según lo detectado por Metaphlan4 y Kraken2, respectivamente. El género Moraxella, representado por las especies Moraxella bovis y Moraxella ovis, también estuvo presente en un número significativo de muestras (hasta el 50% de las muestras según los resultados de Kraken2), sus números alcanzaron el 29% del total de lecturas clasificadas. Sin embargo, los géneros bacterianos detectados utilizando ambos algoritmos de clasificación de lectura, que se presentaron en números bajos (menos del 1 %) o se detectaron en una sola muestra, también incluyeron géneros de patógenos oportunistas como Treponema, Acinetobacter, Mammaliicoccus, Acinetobacter, Micrococcus, Bibersteinia, Proteus, etc. (Figura 3).
La representación de las bacterias detectadas utilizando solo uno de los tres algoritmos en la mayoría de los casos no superó una media del 1% del número total de lecturas clasificadas. Al mismo tiempo, varias de estas especies también fueron descritas como patógenos oportunistas (Finegoldia, Malassezia, etc.). Los bins metagenómicos obtenidos por ensamblaje de novo incluyeron un género no cultivado de la familia Aerococcaceae, a saber, el género Aerococcaceae no cultivado DSM-111022, que está incluido en la base de datos GTDB (71), pero está ausente en las bases de datos clásicas Kraken y Metaphlan, y como resultado no se detectó mediante la clasificación directa de lecturas. Sin embargo, todos los genomas metagenómicos de este género representado en GTDB se recogieron a partir de muestras de leche cruda,1 indicando la validez de este resultado.
3.3 Resultados de ensamblaje de novo y análisis filogenético de bins metagenómicos de alta calidad
Debido a la ubicación distante de los sitios de muestreo y a la diversidad de cepas observada, realizamos un ensamblaje de novo para cada muestra individualmente. Sin embargo, debido a los niveles inconsistentes de contaminación del material metagenómico con el ADN genómico del huésped, la calidad y el tamaño de los ensamblajes variaron considerablemente entre las muestras (Tabla 4). No obstante, seleccionamos los ensamblajes de alta calidad para la agrupación metagenómica. Como resultado, se obtuvieron 10 bins de calidad, suficientes para obtener su clasificación taxonómica utilizando GTDB-toolkit (Tabla 5). Se generaron cuatro bins metagenómicos de alta calidad (completitud >85%, contaminación <5%) a través del binning metagenómico y posteriormente se utilizaron para un análisis filogenético completo.
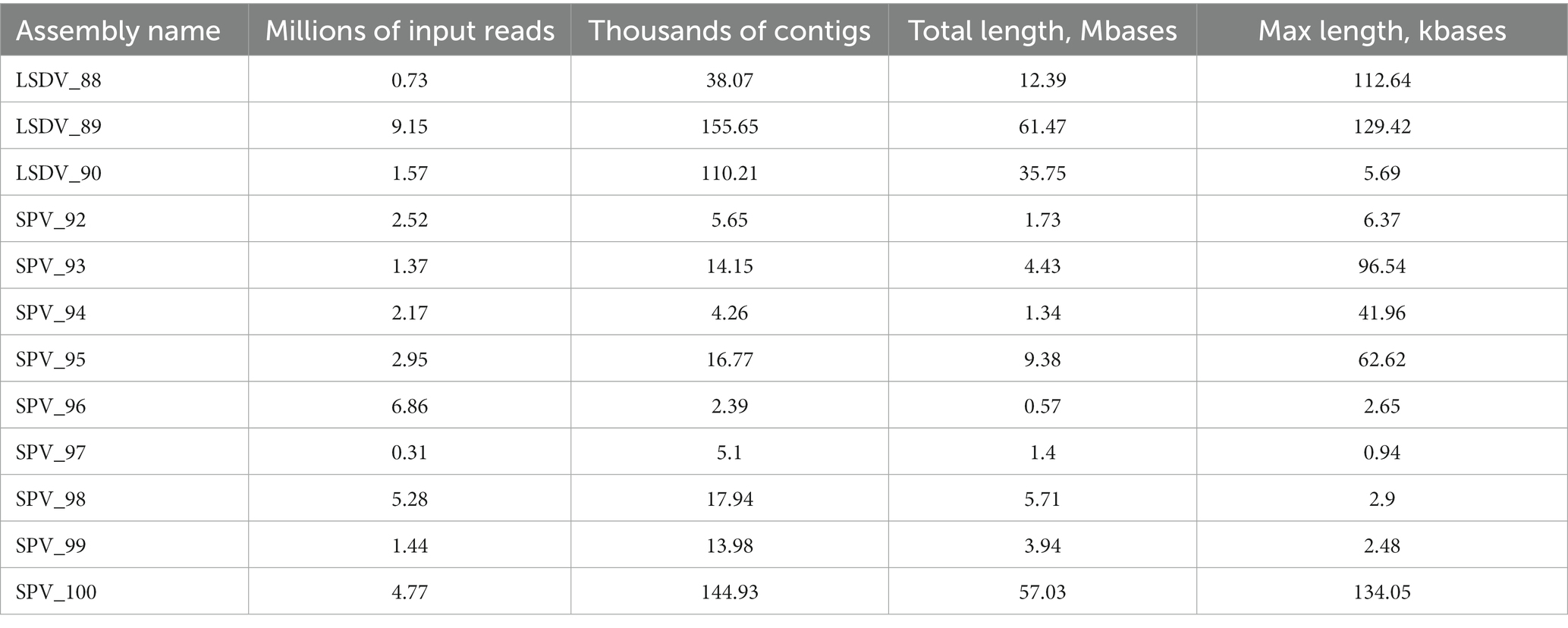 Tabla 4. Resultados del ensamblaje de novo de lecturas metagenómicas.
Tabla 4. Resultados del ensamblaje de novo de lecturas metagenómicas.
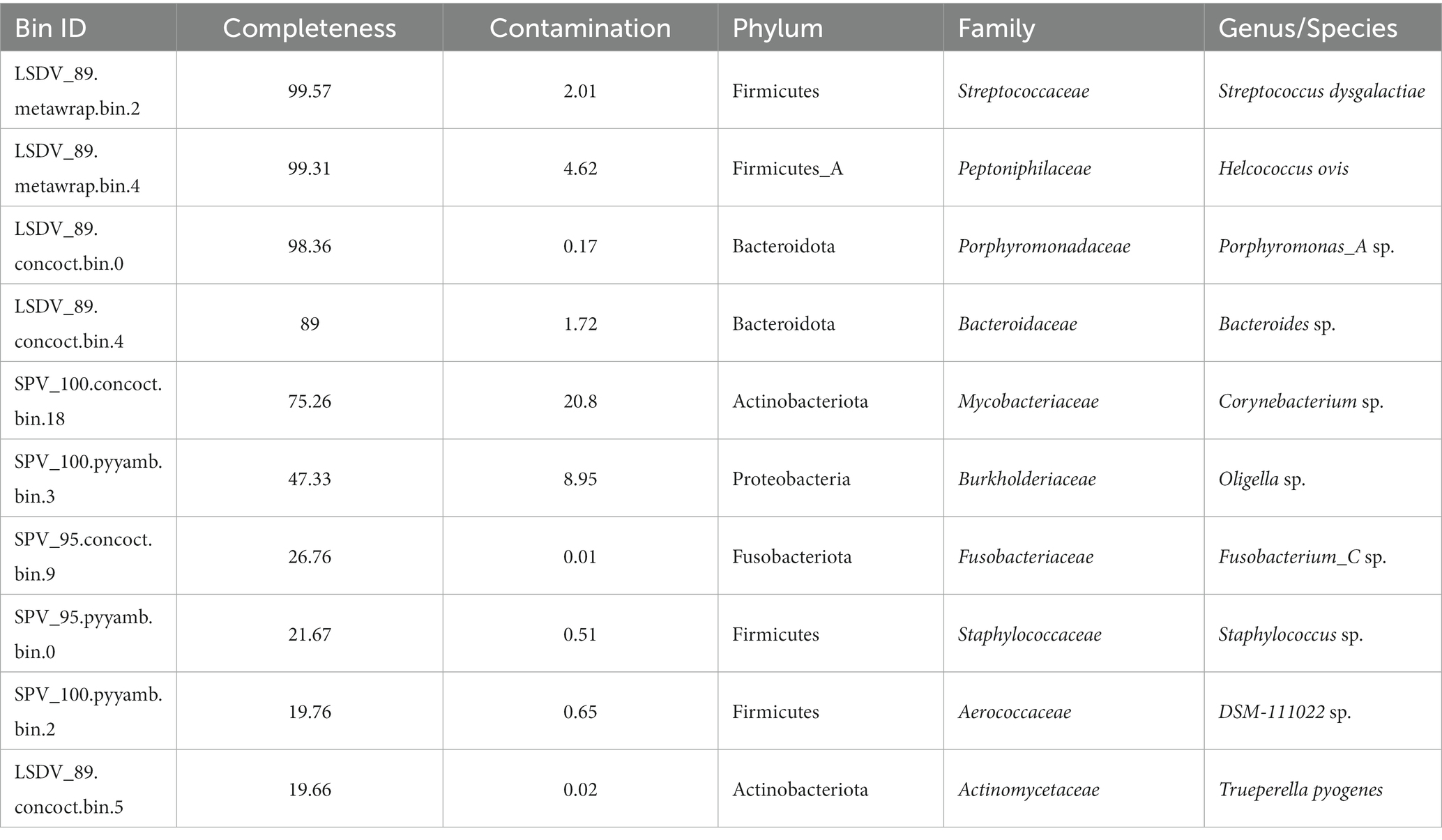 Tabla 5. Obtención de contenedores metagenómicos bacterianos.
Tabla 5. Obtención de contenedores metagenómicos bacterianos.
Para el análisis filogenético basado en secuencias de aminoácidos de 41 genes marcadores se utilizó Bin, clasificado como Helcococcus ovis, con un 99,31% de completitud y un 4,62% de contaminación (Tabla 4). El análisis filogenético confirmó la clasificación inicial; sin embargo, mostró una ligera divergencia con otros genomas de H. ovis aislados de la vagina de bovinos hembras con metritis (72) (Figura suplementaria 3). Otro MAG de alta calidad se clasificó como Streptococcus dysgalactiae, también detectado en grandes cantidades mediante enfoques de clasificación de lectura directa. Dado que hay un gran número de genomas de Streptococcus dysgalactiae en la base de datos del NCBI (185 ensamblajes de genomas en RefSeq a los que se accedió el 01.09.2023), se utilizó un enfoque de tipificación de secuencias multilocus para obtener una filogenia de alta resolución de este contenedor metagenómico. Este contenedor metagenómico se agrupó con los genomas de S. dysgalactiae subespecie dysgalactiae (73) obtenidos en dos estudios a gran escala dirigidos a investigar la filogenia y los factores de virulencia de las cepas de S. dysgalactiae asociadas con la mastitis bovina. En estos dos estudios, uno en Estados Unidos (74) y otro en China (inédito),2 Se aislaron cepas de S. dysgalactiae de la leche de vacas con mastitis. Todas las demás secuencias genómicas de este conglomerado, excepto las ensambladas a partir del material tipo, también se asociaron con la mastitis bovina (Figura suplementaria 4).
Un nuevo género, recientemente reclasificado del género Porphyromonas clásico Porphyromonas_A por el enfoque de taxonomía basada en el genoma (71), fue colocado a través de una reconstrucción filogenética de novo muy cerca de la especie válida Porphyromonas bennonis, aislada de especímenes clínicos humanos, como quistes y abscesos (75 ). Otras especies de referencia de GTDB derivadas del metagenoma, estrechamente relacionadas con este contenedor metagenómico, se ensamblaron a partir de especímenes de microbioma de mamíferos (Figura suplementaria 5). El análisis del cuarto contenedor metagenómico de alta calidad perteneciente al género Bacteroides (89% de completitud y 1,72% de contaminación) mediante la comparación de la identidad promedio de nucleótidos con otros miembros del género (Figura suplementaria 6) indicó su relación más estrecha con las especies de Bacteroides heparynolyticus vinculadas a la inflamación periodontal humana (76). Sin embargo, debe señalarse que el valor de ANI entre LSDV_89.concoct.bin.4 y el genoma de B. heparynolyticus fue de solo 92.26%, lo cual es sustancialmente menor que el valor intraespecie comúnmente reconocido (71, 77). Sin embargo, la calidad de este MAG no es suficiente para proponer una nueva especie de Bacteroides.
3.4 Análisis de genes de resistencia a antibióticos en MAGs metagenómicos obtenidos
La búsqueda de genes de resistencia a antibióticos en MAGs identificados realizada con el paquete staramr (53) mostró solo un grupo de resistencia a tetraciclina tet(W) en el contenedor metagenómico, correspondiente a Oligella no identificada (Tabla 5).
4 Discusión
4.1 Identificación de coinfecciones mediante abordaje metagenómico
El perfil metagenómico de las coinfecciones se ha introducido recientemente en la práctica clínica como resultado de la disminución del coste de las tecnologías de secuenciación de nueva generación (NGS) en la última década (78). El análisis de NGS proporciona datos sin precedentes a nivel pangenómico, lo que brinda la oportunidad de analizar todos los patógenos potenciales en un conjunto de datos (79, 80). El beneficio clave del análisis metagenómico de escopeta, en contraste con el análisis de la composición de la comunidad microbiana utilizando genes marcadores (ARNr 16S), es que permite la detección simultánea de todo tipo de patógenos, incluidos virus, bacterias y microorganismos eucariotas (81). Al mismo tiempo, el alto costo de la secuenciación de escopeta no la convierte en un método universal para el cribado de coinfecciones. Además del hecho de que el análisis de metagenomas completos requiere la obtención de un gran número de lecturas, el ADN de las comunidades microbianas/virales siempre está contaminado con ADN del huésped, con niveles de contaminación que a menudo alcanzan el 95-99% (82). En este contexto, es crucial elegir métodos de análisis de datos que equilibren dos requisitos clave: alta sensibilidad para detectar microorganismos de baja representación y alta especificidad para minimizar los falsos positivos.
En nuestro estudio, se emplearon tres métodos diferentes: examinar la composición de las comunidades microbianas de las costras/lesiones cutáneas de animales infectados por SPV e infectados por LSDV. La clasificación de lecturas utilizando Kraken2 predijo la presencia del mayor número de grupos taxonómicos en la comunidad: se identificaron 172 géneros microbianos (Figura 3). Este resultado se alinea con estudios de referencia anteriores (83, 84) que han encontrado que Kraken 2 es el algoritmo de clasificación más sensible utilizado en la investigación metagenómica. Sin embargo, incluso durante las primeras etapas del análisis, se observó que este algoritmo puede producir resultados falsos positivos. Así, empleando la base de datos estándar extendida Kraken2, se predijo la presencia del patógeno altamente virulento Clostridium botulinum en prácticamente todas las muestras alcanzando el 80% de abundancia. Sin embargo, el análisis de ensamblajes de novo y genes marcadores no pudo confirmar la presencia del patógeno. Un examen más detallado de las lecturas atribuidas a C. botulinum demostró que se originaron a partir de genomas de mamíferos (datos no mostrados). La inclusión de los genomas del huésped en la base de datos personalizada de Kraken, como se describe en la sección Métodos, confirmó que la presencia de C. botulinum era un falso positivo. Otro resultado falso positivo generado por Kraken2 implica la detección de un número significativo de lecturas relacionadas con el patógeno eucariota Babesia bigemina (Figura 2). Sin embargo, este resultado también careció de apoyo por parte de otros métodos. Después de un análisis adicional, se descubrió que ninguna de las lecturas relacionadas con Babesia coincide con ningún cromosoma o andamio largo. En cambio, surgen de dos contigs cortos que parecen haber sido el resultado de la contaminación del genoma de B. bigemina, depositado en el NCBI. Vale la pena señalar que esta observación no está influenciada por el sesgo del algoritmo. En cambio, se debe a una inspección insuficiente y a la purga de la contaminación, incluso de la base de datos RefSeq.
Aparentemente, el examen de la existencia de genes marcadores específicos del clado a través del mapeo directo de lecturas a sus secuencias de nucleótidos puede contrarrestar los sesgos que vienen con la asignación errónea de k-mers a un genoma específico y con la contaminación de genomas disponibles públicamente en bases de datos. De hecho, el análisis realizado con Metaphlan4, que permitió la detección de 54 géneros en un conjunto de datos completo, indicó que la mayoría de los microorganismos eran significativamente abundantes o aparecían en un gran número de muestras. Debido a sus requisitos computacionales comparativamente más bajos, se puede suponer razonablemente que Metaphlan4 es el método de clasificación de lectura directa preferido para aplicaciones clínicas en comparación con Kraken2. Vale la pena mencionar que Metaphlan genera abundancia taxonómica mediante el mapeo de lecturas a genes marcadores de copia única. El número de lecturas asignadas corresponde al número de copias del genoma de la muestra. Por otro lado, los resultados de Kraken2/Bracken no están normalizados por el tamaño del genoma y, por lo tanto, muestran abundancia de secuencias, que representan la cantidad de ADN de un organismo en particular en la muestra (85). Los resultados del perfil de Kraken pueden distorsionar en gran medida la representación real relativa a la proporción de microorganismos que poseen tamaños de genoma muy diferentes, por ejemplo, microorganismos eucariotas y procariotas, o bacterias y virus. En escenarios que involucran comunidades microbianas complejas que consisten en diversos virus, pro- y eucariotas, es casi imposible convertir de manera confiable los valores de abundancia de secuencia en abundancia taxonómica (86). Al estudiar la dinámica de las comunidades microbianas, es crucial rastrear con precisión las relaciones entre varios (micro)organismos. Por lo tanto, el método recomendado es utilizar algoritmos que se basan en el mapeo de lecturas a genes marcadores. El objetivo principal de este estudio es caracterizar las comunidades microbianas de las costras, centrándose en los microbios dominantes. En este sentido, es crucial eliminar los falsos positivos y negativos, por lo que el uso de dos algoritmos alternativos para clasificar las lecturas y comparar los resultados proporciona el enfoque más fiable.
Sin embargo, debe tenerse en cuenta que una limitación significativa de Metaphlan es su dependencia del microbioma humano para formar su base de datos (34). Aunque puede tener una aplicabilidad generalizada entre los mamíferos, su eficacia con otros organismos es incierta y puede producir resultados falsos negativos (56).
El ensamblaje de novo, la agrupación y la adquisición de genomas ensamblados por metagenoma (MAG) de alta calidad son, sin duda, el estándar de oro del análisis metagenómico. No obstante, la generación de MAG de alta calidad requiere una profundidad de secuenciación sustancial, así como tener múltiples muestras del mismo entorno para la agrupación de cobertura diferencial. Desafortunadamente, en el caso del análisis metagenómico de muestras clínicas, esto suele ser imposible, ya que las muestras de diferentes individuos y ubicaciones geográficas no se pueden combinar debido a las diferencias específicas de la cepa. Incluso las diferencias menores en las secuencias del genoma de una especie pueden dar lugar a una calidad de ensamblaje inadecuada y, en consecuencia, a resultados inciertos. En el futuro, el progreso en tecnologías de lectura larga, como Oxford Nanopore, podría resolver este problema. Utilizando el ensamblaje híbrido, se pueden obtener contigs más largos y MAG de alta calidad sin necesidad de agrupamiento a través de la cobertura diferencial (87). Sin embargo, en este trabajo el montaje de novo no dio los resultados esperados. En primer lugar, debido a los elevados niveles de contaminación del ADN del huésped y, por lo tanto, al número insuficiente de lecturas objetivo, el ensamblaje resultó estar fragmentado y la longitud de los contigs fue insuficiente para una clasificación taxonómica fiable o la agrupación. En segundo lugar, la agrupación diferencial no era factible, ya que solo se utilizaba una única muestra por conjunto. Sin embargo, adquirimos 10 bins metagenómicos y logramos establecer su posición taxonómica a través de la taxonomía genómica. De estos, cuatro eran bins de alta calidad, y establecimos con precisión su posición taxonómica y su relación con cepas oportunistas utilizando métodos clásicos de filogenia como la filogenia basada en genes marcadores, MLST y análisis ANI.
Los resultados de este estudio revelan que, aunque Kraken2 cuenta con una alta sensibilidad, la prevalencia de falsos positivos plantea dudas sobre su viabilidad en entornos prácticos. Sin embargo, hay que tener en cuenta que, aunque Metaphlan4 cuenta con una alta especificidad, puede tener dificultades para detectar microorganismos poco representados. Además, la especificidad de la base de datos de genes marcadores utilizada por Metaphlan en relación con el microbioma humano presenta un desafío en su aplicación a la medicina veterinaria. A su vez, el ensamblaje de novo es el método más confiable y específico, sin embargo, requiere una cantidad significativa de datos de secuenciación. Hasta cierto punto, este problema puede resolverse mediante la eliminación del ADN del huésped antes de la secuenciación, ya sea mediante la lisis celular diferencial o mediante la eliminación del ADN de mamíferos mediante la proteína de unión a metilcitosina (88). Al mismo tiempo, una clara ventaja del ensamblaje de novo es la posibilidad de predecir con precisión la resistencia de las cepas oportunistas a los antibióticos, lo que permite la elección adecuada de la terapia antibiótica.
Si bien se están desarrollando activamente enfoques metagenómicos para la investigación veterinaria, actualmente se carece de directrices prácticas para la estandarización del análisis bioinformático (89). Por lo tanto, los investigadores deben decidir sobre la sensibilidad y especificidad de los algoritmos utilizados, en función de la calidad y cantidad de datos primarios en el contexto de los objetivos de investigación. Un factor igualmente importante a la hora de elegir algoritmos y bases de datos es la accesibilidad de la potencia de cálculo, en particular la cantidad de RAM. De acuerdo con la literatura, se puede lograr una sensibilidad y especificidad óptimas mediante el empleo de bases de datos de nucleótidos que abarcan todos los reinos vivos, lo que exige una gran cantidad de memoria (56). Desafortunadamente, en la actualidad, no se puede proporcionar una solución universal para el análisis bioinformático de muestras metagenómicas clínicas. Sin embargo, con base en los datos y resultados del análisis presentados aquí, sugerimos el esquema de toma de decisiones ilustrado en la Figura Suplementaria 6.
4.2 Composición taxonómica de las comunidades microbianas de la sarna
En este estudio caracterizamos la composición de la comunidad microbiana de las costras desarrolladas en las infecciones relacionadas con el capripoxvirus y evaluamos la presencia de patógenos oportunistas. Cabe señalar que, aunque varios estudios han investigado el microbioma y su posible contribución al desarrollo de coinfecciones para diversas enfermedades bovinas (90, 91), la mayoría se han realizado en entornos experimentales controlados y nunca con infecciones por capripoxvirus. Por lo tanto, este estudio, que analiza el microbioma de las lesiones cutáneas en infecciones virales de animales de granja utilizando muestras clínicas reales, es uno de los primeros de este tipo realizado mediante secuenciación de escopeta (92). Desafortunadamente, debido a los estrictos criterios de selección para las condiciones de la muestra y a los desafíos logísticos para obtener muestras clínicas de regiones remotas, el número de muestras involucradas en este estudio fue limitado. Por lo tanto, los autores reconocen que este estudio es un piloto, que proporciona información inicial sobre la diversidad microbiana de las lesiones cutáneas. Sin embargo, no permite una evaluación cuantitativa de los cambios en el microbioma. Además, no es posible comparar los datos obtenidos con otros estudios sobre el microbioma de las lesiones cutáneas en animales de granja con infecciones por capripoxvirus debido a la falta de información al respecto. Por lo tanto, aunque los datos obtenidos en este estudio son muy valiosos, es necesario realizar estudios más extensos, incluyendo estudios longitudinales, para analizar con precisión la diversidad y dinámica de las comunidades microbianas de las costras cutáneas y su papel en la patogénesis de las infecciones por capripoxvirus.
La investigación de la diversidad alfa y beta de la comunidad microbiana en costras/lesiones reveló que, a pesar de las diferencias establecidas entre el SPV y el LSDV (93), así como el microbioma cutáneo de sus respectivos huéspedes (94, 95), no se identificaron diferencias sustanciales entre el LSDV o las lesiones relacionadas con el SPV. Sin embargo, los valores del índice de diversidad fueron inferiores a los reportados en la literatura para una piel sana (59). Esto sugiere que la composición de las comunidades microbianas específicas de las costras formadas durante la infección por capripoxvirus está influenciada predominantemente por uno o dos microorganismos oportunistas predominantes. Cabe señalar que, dado que varios estudios han informado de una asociación de la diversidad alfa de las comunidades microbianas de los órganos afectados por la enfermedad con la respuesta inmunitaria y la gravedad de la enfermedad (96, 97), el análisis de la diversidad alfa en un conjunto de datos más amplio en comparación con las muestras de control de sitios de piel sanos puede proporcionar información adicional sobre los mecanismos del desarrollo de síntomas graves.
Como lo demuestra la clasificación de lectura directa, el ADN viral fue la fuente predominante de las lecturas de secuenciación que no estaban relacionadas con el ADN del huésped, alcanzando el 92% de ellas (Figura 2). Además, teniendo en cuenta los sesgos en la abundancia de secuencias basadas en Kraken2 asociadas con el pequeño tamaño del genoma de los capripoxvirus, podemos suponer que el verdadero número de copias de genomas virales es absolutamente prevalente en la comunidad. En un solo caso, se identificó la coinfección del SPV con el virus orf. El virus Orf se encuentra comúnmente como una coinfección en varias patologías virales (98, 99), aunque su impacto exacto en el desarrollo de los síntomas sigue siendo incierto. A su vez, hubo una gran variación en la composición de las comunidades bacterianas que se desarrollan en muestras de ovejas y vacas. El análisis bioinformático de la parte procariota de las comunidades de la sarna indicó que el patrón de los taxones predominantes es altamente flexible. Es probable que esto se deba a la variabilidad de las bacterias comensales dominantes en un área específica de la piel. No obstante, el análisis de los miembros de la comunidad central identificó pocos géneros detectados en cantidades sustanciales en al menos la mitad de las muestras examinadas (Tabla 3).
La coinfección más prevalente identificada fue Fusobacterium necrophorum, que se sabe que causa varias afecciones graves en el ganado, como absceso en el pie, absceso hepático bovino y difteria en terneros (100). Otro patógeno predominante fue Streptococcus dysgalactiae, que también se confirmó a través del ensamblaje metagenómico de novo y el agrupamiento, se encontró en casi todas las muestras. Esta bacteria tiene el potencial de causar infecciones oportunistas tanto en animales como en humanos en varios sitios del cuerpo (63, 101). Una investigación exhaustiva de la posición filogenética del microorganismo utilizando MLST demostró que la cepa identificada pertenece al mismo grupo que los genomas de S. dysgalactiae obtenidos de leche de vaca con mastitis (74). Este hecho, junto con la abundante evidencia que relaciona a S. dysgalactiae con la mastitis (74, 102), indica que los síntomas de mastitis descritos en ciertas infecciones por capripoxvirus (103) son causados por este patógeno oportunista. Lo mismo puede decirse de otra bacteria, Helcococcus ovis, asociada con metritis en bovinos y ovinos (66, 104). Existe evidencia que sugiere que la metritis se desarrolla con frecuencia como una complicación de la infección por LSDV (105, 106). Por lo tanto, es probable que, al igual que en el caso de S. dysgalactiae, el virus no sea el agente causal directo de este síntoma, sino la coinfección bacteriana.
Trueperella pyogenes, que estaba presente en cantidades relativamente pequeñas pero se detectó en más de la mitad de las muestras, también es un comensal común que causa una variedad de infecciones purulentas en el ganado (107). La correlación entre la presencia de este patógeno y F. necrophorum durante el desarrollo de la enfermedad de la podredumbre del pie en ovejas, descrita por Usie et al. (91), sugiere el desarrollo de un complejo proceso coinfeccioso en las costras.
Otros microorganismos comunes que se encuentran en un número considerable de muestras pertenecen a los géneros Bacteroides y Porphyromonas_A, según la taxonomía GTDB (71). Sin embargo, el análisis filogenético de sus MAG sugiere que pueden pertenecer a especies nuevas y no reportadas de dichos géneros. Por lo tanto, aún no se ha determinado su posible contribución a la aparición de los síntomas resultantes de la infección por SPV/LSDV.
En conjunto, los resultados de este estudio sugieren que las costras desarrollan un proceso activo de coinfección debido a patógenos oportunistas. Es probable que la presencia de coinfección bacteriana sea responsable de síntomas secundarios graves asociados con la infección por LSDV/SPV. Esto concuerda, en parte, con la investigación realizada por Usie et al., quienes utilizaron la metagenómica de escopeta de lesiones cutáneas para revelar la naturaleza multibacteriana de la podredumbre de las ovejas (91). Cabe destacar que en el mencionado estudio se analizó el metagenoma en condiciones experimentales, lo que permitió evaluar la dinámica del proceso. Sin embargo, la presente investigación implicó el análisis directo de muestras clínicas, lo que llevó a discusiones sobre la viabilidad del enfoque metagenómico de escopeta en el entorno clínico. Cabe destacar que la búsqueda de genes de resistencia a antibióticos en los MAGs adquiridos sugiere que estas cepas deberían ser susceptibles a la terapia antibiótica. Este hallazgo apoya la justificación para implementar un tratamiento antibiótico complementario en casos de infección por capripoxvirus.
Al mismo tiempo, el autor debe enfatizar las limitaciones de este estudio debido a su diseño y temática. El uso de muestras clínicas recogidas en condiciones reales de campo permite maximizar la aproximación de las condiciones experimentales a las condiciones prácticas en las que trabajan los veterinarios. Sin embargo, debido a la ausencia de grupos de comparación (excepto para agrupar las muestras según el agente infeccioso) y a la falta de muestreo continuo durante la progresión de la enfermedad, es imposible realizar un análisis funcional del metagenoma y un análisis dinámico de los cambios en la microbiota. Sin embargo, los hallazgos presentados proporcionan una base prometedora para futuras investigaciones sobre la patogénesis de las infecciones relacionadas con el capripoxvirus.
Declaración de disponibilidad de datos
Los archivos de lectura en bruto y los MAG de alta calidad se depositaron en DBJ/EMBL/GenBank bajo el PRJNA1023210 del Bioproyecto.
Declaración ética
No se requirió aprobación ética para el estudio con animales de acuerdo con la legislación local y los requisitos institucionales porque este estudio se basó únicamente en muestras de costra de piel enviadas a FGBI ARRIAH para la confirmación de la infección por capripoxvirus, por lo que no se realizaron experimentos con animales como parte de este trabajo.
Contribuciones de los autores
FS: Curación de datos, Investigación, Software, Visualización, Redacción – borrador original, Redacción – revisión y edición. AM: Investigación, Metodología, Recursos, Redacción – borrador original, Redacción – revisión y edición. AOK: Investigación, Metodología, Recursos, Redacción – Revisión y Edición. OB: Investigación, Metodología, Recursos, Redacción – Revisión y Edición. LP: Administración de proyectos, Recursos, Redacción, revisión y edición. IC: Conceptualización, Obtención de fondos, Redacción – revisión y edición. UZ: Redacción – revisión y edición, curación de datos, investigación, software. ADK: Curación de datos, Software, Escritura – revisión y edición. ASK: Investigación, Metodología, Redacción – revisión y edición. EG: Redacción – revisión y edición, Administración de proyectos, Recursos. AP: Redacción – revisión y edición, Investigación, Metodología. AAK: Redacción – revisión y edición, curación de datos, software. MP: Redacción, revisión y edición, adquisición de fondos, administración de proyectos. ZN: Redacción – revisión y edición. AS: Conceptualización, Supervisión, Redacción – borrador original, Redacción – revisión y edición, Obtención de fondos, Investigación, Administración de proyectos. ST: Conceptualización, Curación de datos, Metodología, Software, Supervisión, Visualización, Redacción – borrador original, Redacción – revisión y edición.
Financiación
El/los autor/es declara(n) haber recibido apoyo financiero para la investigación, autoría y/o publicación de este artículo. Este trabajo fue apoyado por la subvención no. 075-15-2021-1054 del Ministerio de Educación y Ciencia de Rusia para implementar los objetivos del Programa Federal Científico y Técnico para el Desarrollo de tecnologías genéticas durante 2019-2027.
Conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un posible conflicto de intereses.
El/los autor/es declararon, en el momento de la presentación, ser miembro del consejo editorial de Frontiers. Esto no tuvo ningún impacto en el proceso de revisión por pares ni en la decisión final.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o afirmación que pueda ser hecha por su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fvets.2024.1321202/full#supplementary-material
Notas
1. ^https://gtdb.ecogenomic.org/genome?gid=GCA_015863195.1
2. ^https://www.ncbi.nlm.nih.gov/bioproject/PRJNA753266
Referencias
1. Acceso en línea al Código Terrestre . OMSA – Organización Mundial de Sanidad Animal (2023). Disponible en: https://www.woah.org/en/what-we-do/standards/codes-and-manuals/terrestrial-code-online-access/ [Consultado el 7 de octubre de 2023].
2. Hamdi, J , Munyanduki, H , Omari Tadlaoui, K , El Harrak, M y Fassi Fihri, O . Infecciones por capripoxvirus en rumiantes: una revisión. Microorganismos. (2021) 9:902. doi: 10.3390/microorganismos9050902
Resumen de PubMed | Texto completo de Crossref | Google Académico
3. Tulman, ER , Afonso, CL , Lu, Z , Zsak, L , Sur, J-H , Sandybaev, NT, et al. Los genomas de la viruela ovina y los virus de la viruela caprina. J Virol. (2002) 76:6054–61. doi: 10.1128/JVI.76.12.6054-6061.2002
Resumen de PubMed | Texto completo de Crossref | Google Académico
4. Whittle, L , Chapman, R y Williamson, A-L . Enfermedad de la piel nodular contagiosa: una enfermedad emergente del ganado en Europa y Asia. Vacuna. (2023) 11:578. doi: 10.3390/vacunas11030578
Resumen de PubMed | Texto completo de Crossref | Google Académico
5. Brennan, G , Stoian, AMM , Yu, H , Rahman, MJ , Banerjee, S , Stroup, JN, et al. Mecanismos moleculares de la evolución de los poxvirus. MBio. (2023) 14:e0152622. doi: 10.1128/mbio.01526-22
Resumen de PubMed | Texto completo de Crossref | Google Académico
6. Sprygin, A , Mazloum, A , van Schalkwyk, A y Babiuk, S . Capripoxvirus, leporipoxvirus y orthopoxvirus: ocurrencias de recombinación. Microbiol frontal. (2022) 13. doi: 10.3389/fmicb.2022.978829
Resumen de PubMed | Texto completo de Crossref | Google Académico
7. Dao, TD , Tran, LH , Nguyen, HD , Hoang, TT , Nguyen, GH , Tran, KVD, et al. Caracterización del virus de la dermatosis nodular contagiosa aislado de una jirafa en Vietnam. Transbound Emerg Dis. (2022) 69:E3268–72. doi: 10.1111/tbed.14583
Resumen de PubMed | Texto completo de Crossref | Google Académico
8. Tuppurainen, MEDE , Venter, EH , Shisler, JL , Gari, G , Mekonnen, GA , Juleff, N, et al. Revisión: Enfermedades por capripoxvirus: estado actual y oportunidades para el control. Transbound Emerg Dis. (2017) 64:729–45. doi: 10.1111/tbed.12444
Resumen de PubMed | Texto completo de Crossref | Google Académico
9. Babiuk, S , Bowden, TR , Boyle, DB , Wallace, DB y Kitching, RP . Capripoxvirus: una amenaza mundial emergente para el ganado ovino, caprino y bovino. Transbound Emerg Dis. (2008) 55:263–72. doi: 10.1111/j.1865-1682.2008.01043.x
Resumen de PubMed | Texto completo de Crossref | Google Académico
10. Zeynalova, S , Asadov, K , Guliyev, F , Vatani, M y Aliyev, V . Epizootología y diagnóstico molecular de la dermatosis nodular contagiosa en el ganado de Azerbaiyán. Microbiol frontal. (2016) 7:1022. doi: 10.3389/fmicb.2016.01022
Resumen de PubMed | Texto completo de Crossref | Google Académico
11. Babiuk, S , Bowden, TR , Parkyn, G , Dalman, B , Manning, L , Neufeld, J, et al. Cuantificación del virus de la dermatosis nodular contagiosa tras la infección experimental en bovinos. Transbound Emerg Dis. (2008) 55:299–307. doi: 10.1111/j.1865-1682.2008.01024.x
Resumen de PubMed | Texto completo de Crossref | Google Académico
12. Nesterov, A , Mazloum, A , Byadovskaya, O , Shumilova, I , Van Schalkwyk, A , Krotova, A, et al. Un estudio controlado experimentalmente indica que la cepa natural de la enfermedad cutánea nodular contagiosa recombinante similar a una vacuna Udmurtiya/2019, detectada durante el invierno helado en latitudes septentrionales, se transmite por contacto indirecto. Front Vet Sci. (2022) 9:1001426. doi: 10.3389/fvets.2022.1001426
Resumen de PubMed | Texto completo de Crossref | Google Académico
13. Haegeman, A , Sohier, C , Mostin, L , De Leeuw, I , Van Campe, W , Philips, W, et al. Evidencia de transmisión del virus de la dermatosis nodular contagiosa en bovinos infectados subclínicamente por Stomoxys calcitrans. Virus. (2023) 15:1285. doi: 10.3390/v15061285
Resumen de PubMed | Texto completo de Crossref | Google Académico
14. Sanz-Bernardo, B , Haga, IR , Wijesiriwardana, N , Basu, S , Larner, W , Díaz, AV, et al. La cuantificación y modelización de la adquisición y retención del virus de la dermatosis nodular contagiosa por insectos hematófagos revela que el ganado afectado clínicamente, pero no subclínicamente, es promotor de la transmisión viral y objetivos clave para el control de brotes de enfermedades. J Virol. (2021) 95:e02239–20. doi: 10.1128/JVI.02239-20
Resumen de PubMed | Texto completo de Crossref | Google Académico
15. Shumilova, I , Sprygin, A , Mazloum, A , Pronin, V , Byadovskaya, O , Babiuk, S, et al. Comparación de la patología macroscópica entre los virus de la dermatosis nodular contagiosa clásica y recombinante. Virus. (2023) 15:1883. doi: 10.3390/v15091883
16. Nagar, P y Hasija, Y . Enfoque metagenómico en el estudio y tratamiento de diversas enfermedades de la piel: una breve revisión. Biomed Dermatol. (2018) 2:19. doi: 10.1186/s41702-018-0029-4
17. Hajer, I , Abbas, B y Abu Samra, MT . Virus Capripox en ovejas y cabras en Sudán. Rev Elev Med Vet Pays Trop. (1988) 41:125–8. doi: 10.19182/remvt.8711
Resumen de PubMed | Texto completo de Crossref | Google Académico
18. Molina-Mora, J.A. , Cordero-Laurent, E , Calderón-Osorno, M , Chacón-Ramírez, E y Duarte-Martínez, F . Pipeline metagenómico para identificar coinfecciones entre distintas variantes preocupantes del SARS-CoV-2: casos de estudio desde alfa hasta ómicron. Sci Rep. (2022) 12:9377. doi: 10.1038/s41598-022-13113-4
Resumen de PubMed | Texto completo de Crossref | Google Académico
19. Zhang, H , Liu, G , Él, L , Zhu, Y , Ma, H y Zhuang, S . Coinfección pulmonar potencialmente mortal con Mycobacterium tuberculosis y aspergillus lentulus en un paciente diabético diagnosticado por secuenciación de nueva generación del metagenoma. BMC infecta dis. (2023) 23:88. doi: 10.1186/s12879-023-08052-y
Resumen de PubMed | Texto completo de Crossref | Google Académico
20. Suminda, GGD , Bhandari, S , Ganó, Y , Goutam, U , Kanth Pulicherla, K , Son, Y-O, et al. Tecnologías de secuenciación de alto rendimiento en la detección de patógenos ganaderos, diagnóstico y vigilancia zoonótica. Comput Struct Biotechnol J. (2022) 20:5378–92. doi: 10.1016/j.csbj.2022.09.028
Resumen de PubMed | Texto completo de Crossref | Google Académico
21. Munang’andu, HM , Mugimba, KK , Byarugaba, DK , Mutoloki, S y Evensen, Ø . Avances actuales en el descubrimiento de virus y el papel diagnóstico de la metagenómica viral en organismos acuáticos. Microbiol frontal. (2017) 8. doi: 10.3389/fmicb.2017.00406
Resumen de PubMed | Texto completo de Crossref | Google Académico
22. Muhamad Rizal, NS , Neoh, H , Ramli, R , Periyasamy, Relaciones Públicas , Hanafiah, A , Abdul Samat, MN, et al. Ventajas y limitaciones de la secuenciación de nueva generación de ARNr 16S para la identificación de patógenos en el laboratorio de microbiología diagnóstica: perspectivas desde un país de ingresos medios. Diagnóstico. (2020) 10:816. doi: 10.3390/diagnostics10100816
Resumen de PubMed | Texto completo de Crossref | Google Académico
23. Wensel, CR , Pluznick, JL , Salzberg, S.L. y Sears, CL . Secuenciación de nueva generación: información para avanzar en las investigaciones clínicas del microbioma. J Clin Invest. (2022) 132. doi: 10.1172/JCI154944
Resumen de PubMed | Texto completo de Crossref | Google Académico
24. Couto, N , Schuele, L , Raangs, CE , Machado, diputado , Mendes, CI , Jesús, TF, et al. Pasos críticos en la metagenómica clínica de escopeta para la detección y tipificación concomitantes de patógenos microbianos. Sci Rep. (2018) 8:13767. DOI: 10.1038/s41598-018-31873-w
Resumen de PubMed | Texto completo de Crossref | Google Académico
25. Bengtsson-Palme, J , Boulund, F , Fick, J , Kristiansson, E y Larsson, DGJ . La metagenómica de escopeta revela una amplia gama de genes de resistencia a antibióticos y elementos móviles en un lago contaminado en la India. Microbiol frontal. (2014) 5:5. doi: 10.3389/fmicb.2014.00648
Resumen de PubMed | Texto completo de Crossref | Google Académico
26. Sprygin, A , Mazloum, A , Van Schalkwyk, A , Krotova, A , Shalina, K , Dmitric, M, et al. El desarrollo de un ensayo de PCR en tiempo real para la detección específica de la cepa de ADN del virus de la vacuna contra la viruela ovina NISKHI. Aplicación Microbiol. (2022) 2:956–64. doi: 10.3390/applmicrobiol2040073
27. Meslier, V , Quinquis, B , Da Silva, K , Plaza Oñate, F , Pons, N , Roume, H, et al. Benchmarking de plataformas de secuenciación de segunda y tercera generación para metagenómica microbiana. Datos de sci. (2022) 9:694. doi: 10.1038/s41597-022-01762-z
Resumen de PubMed | Texto completo de Crossref | Google Académico
28. Shen, W , Le, S , Li, Y y Hu, F . SeqKit: Un kit de herramientas multiplataforma y ultrarrápido para la manipulación de archivos FASTA/Q. PloS Uno. (2016) 11:e0163962. doi: 10.1371/journal.pone.0163962
Resumen de PubMed | Texto completo de Crossref | Google Académico
29. Bioinformática de Babraham . FastQC Una herramienta de control de calidad para datos de secuencia de alto rendimiento (2010). Disponible en: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Consultado el 28 de noviembre de 2022].
30. Chen, S , Zhou, Y , Chen, Y y Gu, J . Fastp: un preprocesador FASTQ todo en uno ultrarrápido. Bioinformática. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
Resumen de PubMed | Texto completo de Crossref | Google Académico
31. Langmead, B y Salzberg, S.L. . Alineación rápida de lectura con espacio con pajarita 2. Métodos Nat. (2012) 9:357–9. doi: 10.1038/nmeth.1923
Resumen de PubMed | Texto completo de Crossref | Google Académico
32. Lu, J , Breitwieser, FP , Thielen, P y Salzberg, S.L. . Bracken: estimación de la abundancia de especies en datos metagenómicos. PeerJ Comput Sci. (2017) 3:E104. doi: 10.7717/peerj-cs.104
33. Lu, J , Rincón, N , Madera, DE , Breitwieser, FP , Pockrandt, C , Langmead, B, et al. Análisis del metagenoma utilizando el paquete de software kraken. Nat Protoc. (2022) 17:2815–39. doi: 10.1038/s41596-022-00738-y
Resumen de PubMed | Texto completo de Crossref | Google Académico
34. Blanco-Míguez, A , Beghini, F , Cumbo, F , McIver, LJ , Thompson, KN , Zolfo, M, et al. Ampliación y mejora de los perfiles taxonómicos metagenómicos con especies no caracterizadas mediante MetaPhlAn 4. Nat Biotechnol. (2023) 41:1633–44. DOI: 10.1038/s41587-023-01688-w
Resumen de PubMed | Texto completo de Crossref | Google Académico
35. Nurk, S. , Meleshko, D. , Korobeynikov, A. y Pevzner, P. (2016). metaSPAdes: Un nuevo ensamblador versátil de metagenómica de novo. Disponible en: https://www.semanticscholar.org/paper/metaSPAdes%3A-A-New-Versatile-de-novo-Metagenomics-Nurk-Meleshko/7e08158ef27cf1732d589e58e807377074b151cb [Consultado el 15 de diciembre de 2022].
36. Página, AJ , De Silva, N , Caza, M , Codorniz, MA , Parkhill, J , Harris, SR, et al. Robusto ensamblaje de procariotas de novo de alto rendimiento y canalización de mejora para los datos de Illumina. Genómica microbiana. (2016) 2:E000083. doi: 10.1099/mgen.0.000083
Resumen de PubMed | Texto completo de Crossref | Google Académico
37. Alneberg, J , Bjarnason, BS , De Bruijn, I , Schirmer, M , Rápido, J , Ijaz, UZ, et al. Agrupación de contigs metagenómicos por cobertura y composición. Métodos Nat. (2014) 11:1144–6. doi: 10.1038/nmeth.3103
Resumen de PubMed | Texto completo de Crossref | Google Académico
38. Kang, DD , Li, F , Kirton, E , Tomás, A , Egan, R , An, H, et al. MetaBAT 2: un algoritmo de agrupamiento adaptativo para la reconstrucción robusta y eficiente del genoma a partir de ensamblajes de metagenomas. PeerJ. (2019) 7:e7359. doi: 10.7717/peerj.7359
Resumen de PubMed | Texto completo de Crossref | Google Académico
39. Wu, Y-W y Singer, SW . Recuperación de genomas individuales a partir de metagenomas utilizando MaxBin 2.0. Protocolos vigentes. (2021) 1:e128. doi: 10.1002/cpz1.128
Resumen de PubMed | Texto completo de Crossref | Google Académico
40. Uritskiy, GV , DiRuggiero, J y Taylor, J . MetaWRAP: una canalización flexible para el análisis de datos metagenómicos resueltos por el genoma. Microbioma. (2018) 6:1–13. doi: 10.1186/s40168-018-0541-1
41. Korzhenkov, A. (2019). YAMB: agrupamiento del metagenoma mediante reducción de dimensionalidad no lineal y agrupamiento basado en la densidad. bioRxiv :521286. doi: 10.1101/521286
42. Sieber, CMK , Probst, AJ , Sharrar, A , Thomas, Columbia Británica , Hess, M , Tringe, SG, et al. Recuperación de genomas a partir de metagenomas a través de una estrategia de desreplicación, agregación y puntuación. Nat Microbiol. (2018) 3:836–43. doi: 10.1038/s41564-018-0171-1
Resumen de PubMed | Texto completo de Crossref | Google Académico
43. Chaumeil, P-A , Mussig, AJ , Hugenholtz, P y Parques, DH . GTDB-Tk: un conjunto de herramientas para clasificar genomas con la Base de Datos de Taxonomía Genómica. Bioinformática. (2020) 36:1925–7. doi: 10.1093/bioinformatics/btz848
Resumen de PubMed | Texto completo de Crossref | Google Académico
44. Alex ChklovskiParks, DH , Woodcroft, BJ y Tyson, GW . CheckM2: una herramienta rápida, escalable y precisa para evaluar la calidad del genoma microbiano mediante el aprendizaje automático. Métodos Nat. (2023) 20:1203–12. DOI: 10.1038/s41592-023-01940-w
Resumen de PubMed | Texto completo de Crossref | Google Académico
45. Mistry, J , Chuguransky, S , Williams, L , Qureshi, M , Salazar, GA , Sonnhammer, ELL, et al. Pfam: la base de datos de familias de proteínas en 2021. Ácidos nucleicos Res. (2021) 49:D412-9. doi: 10.1093/nar/gkaa913
Resumen de PubMed | Texto completo de Crossref | Google Académico
46. Remolino, SR . Búsquedas aceleradas de perfiles HMM. PLoS Comput Biol. (2011) 7:E1002195. doi: 10.1371/journal.pcbi.1002195
Resumen de PubMed | Texto completo de Crossref | Google Académico
47. Katoh, K y Standley, DM . Software de alineación de secuencia múltiple MAFFT versión 7: mejoras en el rendimiento y la usabilidad. Mol Biol Evol. (2013) 30:772–80. DOI: 10.1093/molbev/mst010
Resumen de PubMed | Texto completo de Crossref | Google Académico
48. Precio, MN , Dehal, PS y Arkin, AP . FastTree 2: árboles de máxima probabilidad aproximada para alineaciones grandes. PloS Uno. (2010) 5:e9490. doi: 10.1371/journal.pone.0009490
Resumen de PubMed | Texto completo de Crossref | Google Académico
49. Guerrero-Araya, E , Muñoz, M , Rodríguez, C y Paredes-Sabja, D . FastMLST: Una herramienta multinúcleo para la tipificación de secuencias multilocus de borradores de ensamblajes de genomas. Bioinform Biol Insights. (2021) 15:11779322211059238. doi: 10.1177/11779322211059238
Resumen de PubMed | Texto completo de Crossref | Google Académico
50. Minh, BQ , Schmidt, HA , Chernomor, O , Schrempf, D , Woodhams, MD , von Haeseler, A, et al. IQ-TREE 2: nuevos modelos y métodos eficientes para la inferencia filogenética en la era genómica. Mol Biol Evol. (2020) 37:1530–4. DOI: 10.1093/Molbev/MSAA015
Resumen de PubMed | Texto completo de Crossref | Google Académico
51. Letunic, I y Bork, P . Árbol interactivo de la vida (iTOL) v5: una herramienta en línea para la visualización y anotación de árboles filogenéticos. Ácidos nucleicos Res. (2021) 49:W293–6. doi: 10.1093/nar/gkab301
Resumen de PubMed | Texto completo de Crossref | Google Académico
52. Pritchard, L , Glover, H , Humphris, S , Elphinstone, J , y Toth, yo . Genómica y taxonomía en el diagnóstico para la seguridad alimentaria: patógenos de plantas enterobacterianas en descomposición blanda. Métodos anales. (2016) 8:12–24. doi: 10.1039/C5AY02550H
53. Bharat, A , Petkau, A , Avery, BP , Chen, JC , Folster, JP , Carson, CA, et al. Correlación entre la detección fenotípica e in silico de resistencia antimicrobiana en Salmonella enterica en Canadá mediante Staramr. Microorganismos. (2022) 10:292. DOI: 10.3390/microorganismos10020292
Resumen de PubMed | Texto completo de Crossref | Google Académico
54. Jurasz, H , Pawłowski, T y Perlejewski, K . Problema de contaminación en metagenómica viral: problemas, soluciones y perspectivas clínicas. Microbiol frontal. (2021) 12:745076. doi: 10.3389/fmicb.2021.745076
Resumen de PubMed | Texto completo de Crossref | Google Académico
55. Madera, DE , Lu, J y Langmead, B . Análisis metagenómico mejorado con kraken 2. Genoma Biol. (2019) 20:257. doi: 10.1186/s13059-019-1891-0
Resumen de PubMed | Texto completo de Crossref | Google Académico
56. Wright, RJ , Comeau, AM y Langille, MGI . De los valores predeterminados a las bases de datos: la elección de parámetros y bases de datos tiene un impacto drástico en el rendimiento de las herramientas de clasificación taxonómica metagenómica. Genoma Microb. (2023) 9:mgen000949. doi: 10.1099/mgen.0.000949
Resumen de PubMed | Texto completo de Crossref | Google Académico
57. Gu, Y , Sol, J , Li, K , Wu, X y Zhang, J . Alteración en el microbioma cutáneo en la enfermedad cutánea de injerto contra huésped. Acta Derm Venereol. (2021) 101:adv00374. doi: 10.2340/00015555-3613
Resumen de PubMed | Texto completo de Crossref | Google Académico
58. Khadka, VD , Clave, FM , Romo-González, C , Martínez-Gayosso, A , Campos-Cabrera, BL , Gerónimo-Gallegos, A, et al. El microbioma cutáneo de los pacientes con dermatitis atópica se normaliza gradualmente durante el tratamiento. Las células frontales infectan el microbiol. (2021) 11. doi: 10.3389/fcimb.2021.720674
Resumen de PubMed | Texto completo de Crossref | Google Académico
59. Ekman, L , Bagge, E , Nyman, A , Persson Waller, K , Pringle, M y Segerman, B . Una investigación metagenómica de escopeta de la microbiota de la dermatitis por hendidura de la ubre en comparación con la piel sana de las vacas lecheras. PloS Uno. (2020) 15:e0242880. doi: 10.1371/journal.pone.0242880
Resumen de PubMed | Texto completo de Crossref | Google Académico
60. Spyrou, V y Valiakos, G . Infección por el virus Orf en ovejas o cabras. Microbiol veterinario. (2015) 181:178–82. doi: 10.1016/j.vetmic.2015.08.010
61. Van Metre, DC . Patogenia y tratamiento de la podredumbre del pie bovino. Vet Clin North Am Food Anim Pract. (2017) 33:183–94. doi: 10.1016/j.cvfa.2017.02.003
62. Fentie, T , Fenta, N , Leta, S , Molla, W , Ayele, B , Teshome, Y, et al. Seroprevalencia, factores de riesgo y distribución de la viruela ovina y caprina en la región de Amhara. Etiopía BMC Vet Res. (2017) 13:385. doi: 10.1186/s12917-017-1312-0
Resumen de PubMed | Texto completo de Crossref | Google Académico
63. Zadoks, RN , Middleton, JR , McDougall, S , Katholm, J y Schukken, YH . Epidemiología molecular de los patógenos de mastitis en ganado lechero y relevancia comparativa para los seres humanos. J Neoplasia Biol de la glándula mamaria. (2011) 16:357–72. doi: 10.1007/s10911-011-9236-y
Resumen de PubMed | Texto completo de Crossref | Google Académico
64. Foster, AP . Enfermedad cutánea estafilocócica en el ganado. Veterinario Dermatol. (2012) 23:E63. doi: 10.1111/j.1365-3164.2012.01093.x
65. G Abril, A , G Villa, T , Barros-Velázquez, J , Cañas, B , Sánchez-Pérez, A , Calo-Mata, P, et al. Exotoxinas de Staphylococcus aureus y su detección en la industria láctea y mastitis. Toxinas (Basilea). (2020) 12:537. doi: 10.3390/toxinas12090537
Resumen de PubMed | Texto completo de Crossref | Google Académico
66. Jeon, SJ , Vieira-Neto, A , Gobikrushanth, M , Daetz, R , Mingoti, RD , Parize, ACB, et al. Progresión de la microbiota uterina desde el parto hasta el establecimiento de la metritis en vacas lecheras. Appl Environ Microbiol. (2015) 81:6324–32. doi: 10.1128/AEM.01753-15
Resumen de PubMed | Texto completo de Crossref | Google Académico
67. Dr. Collins , Falsen, E , Foster, G , Monasterio, LR , Domínguez, L y Fernández-Garazabal, JF . Helcococcus ovis sp. nov., un organismo grampositivo de ovejas. Int J Syst Bacteriol. (1999) 49 Pe 4:1429-32. doi: 10.1099/00207713-49-4-1429
Resumen de PubMed | Texto completo de Crossref | Google Académico
68. Zhang, Y , Cui, J , Parkinson, A , Hayes, J , Ott, K y Byrum, B . Aislamiento de Helcococcus ovis de ovejas con pleuritis y bronconeumonía. J Vet Diagn Invest. (2009) 21:164–6. doi: 10.1177/104063870902100130
Resumen de PubMed | Texto completo de Crossref | Google Académico
69. Liu, K , Deng, Z , Zhang, L , Gu, X , Liu, G , Liu, Y, et al. Características biológicas y patogenicidad de Helcococcus ovis aislado de mastitis bovina clínica en un hato lechero chino. Front Vet Sci. (2022) 8:8. doi: 10.3389/fvets.2021.756438
Resumen de PubMed | Texto completo de Crossref | Google Académico
70. Hulsen, T. (2022). DeepVenn: una aplicación web para la creación de diagramas de Venn proporcionales al área utilizando el marco de aprendizaje profundo tensorflow. arXiv. doi: 10.48550/arXiv.2210.04597
71. Parques, DH , Chuvochina, M , Rinke, C , Mussig, AJ , Chaumeil, P-A y Hugenholtz, P . GTDB: un censo continuo de la diversidad bacteriana y arqueológica a través de una taxonomía basada en el genoma filogenéticamente consistente, normalizada y completa. Ácidos nucleicos Res. (2022) 50:D785–94. doi: 10.1093/nar/gkab776
Resumen de PubMed | Texto completo de Crossref | Google Académico
72. Cunha, F , Jeon, SJ , Kutzer, P , Jeong, KC y Galvão, KN . Borrador de secuencias genómicas de cepas de Helcococcus ovis aisladas en el momento del diagnóstico de metritis del útero de vacas lecheras Holstein. Anuncio de Microbiol Resour. (2019) 8:E00402–19. doi: 10.1128/MRA.00402-19
Resumen de PubMed | Texto completo de Crossref | Google Académico
73. Vandamme, P , Olla, B , Falsen, E , Kersters, K y Devriese, LA . Estudio taxonómico de los grupos de estreptococos C, G y L (Streptococcus dysgalactiae) y propuesta de S. dysgalactiae subsp. equisimilis subsp. nov. Int J Syst Bacteriol. (1996) 46:774–81. doi: 10.1099/00207713-46-3-774
Resumen de PubMed | Texto completo de Crossref | Google Académico
74. Crippa, BL , Rodrigues, MX , Tomazi, T , Yang, Y , de Oliveira Rocha, L , Bicalho, RC, et al. Factores de virulencia, resistencia antimicrobiana y filogenia de Streptococcus dysgalactiae asociado a mastitis bovina. J Dairy Res. (2023) 90:152–7. doi: 10.1017/S0022029923000195
Resumen de PubMed | Texto completo de Crossref | Google Académico
75. Summanen, PH , Lawson, PA y Finegold, SM . Porphyromonas bennonis sp. nov., aislado de especímenes clínicos humanos. Int J Syst Evol Microbiol. (2009) 59:1727–32. doi: 10.1099/ijs.0.001909-0
Resumen de PubMed | Texto completo de Crossref | Google Académico
76. Okuda, K , Kato, T , Shiozu, J , Takazoe, I y Nakamura, T . Bacteroides heparinolyticus sp. nov. aislado de humanos con periodontitis. Int J Syst Evol Microbiol. (1985) 35:438–42. doi: 10.1099/00207713-35-4-438
77. Olm, MR , Crits-Christoph, A , Diamante, S , Lavy, A , Matheus Carnevali, PB y Banfield, JF . Métricas consistentes derivadas del metagenoma Verifique y delinee los límites de las especies bacterianas. mSystems. (2020) 5:00731. doi: 10.1128/msystems.00731-19
78. Li, C-X , Li, W , Zhou, J , Zhang, B , Feng, Y , Xu, C-P, et al. Caracterización metagenómica de alta resolución de infeccios complejos en infección respiratoria aguda pediátrica. Sci Rep. (2020) 10:3963. doi: 10.1038/s41598-020-60992-6
Resumen de PubMed | Texto completo de Crossref | Google Académico
79. Mitra, N , Cernicchiaro, N , Torres, S , Li, F y Hause, BM . La caracterización metagenómica del viroma asociado con la enfermedad respiratoria bovina en el ganado de engorde identificó nuevos virus y sugiere un papel etiológico para el virus de la influenza D. J Gen Virol. (2016) 97:1771–84. doi: 10.1099/jgv.0.000492
Resumen de PubMed | Texto completo de Crossref | Google Académico
80. Zhang, M , Colina, JE , Alexander, TW y Huang, Y . Los viromas nasales del ganado a su llegada a los corrales de engorde del oeste de Canadá y su relación con el desarrollo de enfermedades respiratorias bovinas. Transbound Emerg Dis. (2021) 68:2209–18. doi: 10.1111/tbed.13873
Resumen de PubMed | Texto completo de Crossref | Google Académico
81. Durazzi, F , Sala, C , Castellani, G , Manfreda, G , Remondini, D y De Cesare, A . Comparación entre el ARNr 16S y los datos de secuenciación de escopeta para la caracterización taxonómica de la microbiota intestinal. Sci Rep. (2021) 11:3030. doi: 10.1038/s41598-021-82726-y
Resumen de PubMed | Texto completo de Crossref | Google Académico
82. McArdle, AJ y Kaforou, M . Sensibilidad de la metagenómica de escopeta al ADN del huésped: las estimaciones de abundancia dependen de herramientas bioinformáticas y la contaminación es el principal problema. Accede a Microbiol. (2020) 2:acmi000104. doi: 10.1099/acmi.0.000104
Resumen de PubMed | Texto completo de Crossref | Google Académico
83. Gris, M , Zhao, Z y Rosen, GL . ¿Qué tan escalables son los métodos hash basados en K-Mer de marcadores específicos de clado para la clasificación taxonómica metagenómica? Proceso de Señal Frontal. (2022) 2:2513. doi: 10.3389/frsip.2022.842513
84. LaPierre, N , Alser, M , Eskin, E , Koslicki, D y Mangul, S . Metalign: perfil metagenómico eficiente basado en alineaciones a través de hash mínimo de contención. Genoma Biol. (2020) 21:242. doi: 10.1186/s13059-020-02159-0
Resumen de PubMed | Texto completo de Crossref | Google Académico
85. Liu, B , Gibbons, T , Ghodsi, M , Treangen, T y Pop, M . Estimación precisa y rápida de perfiles taxonómicos a partir de secuencias metagenómicas de escopeta. BMC Genómica. (2011) 12:S4. doi: 10.1186/1471-2164-12-S2-S4
Resumen de PubMed | Texto completo de Crossref | Google Académico
86. Sol, Z , Huang, S , Zhang, M , Zhu, Q , Haiminen, N , Carrieri, AP, et al. Desafíos en la evaluación comparativa de los perfiladores metagenómicos. Métodos Nat. (2021) 18:618–26. doi: 10.1038/s41592-021-01141-3
Resumen de PubMed | Texto completo de Crossref | Google Académico
87. Sí, L , Dong, N , Xiong, W , Li, J , Li, R , Heng, H, et al. Metagenómica de alta resolución de la microbiota intestinal humana generada por el ensamblaje del metagenoma híbrido de Nanopore e Illumina. Microbiol frontal. (2022) 13:801587. doi: 10.3389/fmicb.2022.801587
Resumen de PubMed | Texto completo de Crossref | Google Académico
88. Jeltsch, A , Broche, J , Lungu, C y Bashtrykov, P . Aplicaciones biotecnológicas de proteínas de dominio MBD para el análisis de metilación del ADN. J Mol Biol. (2020) 432:1816–23. doi: 10.1016/j.jmb.2019.08.020
89. Brunak, S , Bjerre Collin, C , Eva Ó Cathaoir, K , Golebiewski, M , Kirschner, M , Kockum, I, et al. Hacia directrices de estandarización para enfoques in silico en medicina personalizada. J Integr Bioinform. (2020) 17:20200006. doi: 10.1515/jib-2020-0006
Resumen de PubMed | Texto completo de Crossref | Google Académico
90. Ng, TFF , Kondov, NO , Deng, X , Van Eenennaam, A , Neibergs, HL y Delwart, E . Un estudio de metagenómica y casos y controles para identificar virus asociados con enfermedades respiratorias bovinas. J Virol. (2015) 89:5340–9. doi: 10.1128/jvi.00064-15
Resumen de PubMed | Texto completo de Crossref | Google Académico
91. Usié, A , Leão, C , Gaspar, D , Monteiro, H , Tábuas, L , Bettencourt, E, et al. Un enfoque metagenómico para caracterizar el microbioma de la pudrición del pie en ovejas merinas. Microbiol veterinario. (2023) 281:109745. doi: 10.1016/j.vetmic.2023.109745
Resumen de PubMed | Texto completo de Crossref | Google Académico
92. Hansen, S , Pessôa, R , Nascimento, A , El-Tholoth, M , Abd El Wahed, A y Sanabani, SS . Conjunto de datos de la composición del microbioma en lesiones cutáneas causadas por el virus de la enfermedad cutánea nodular contagiosa mediante secuenciación paralela masiva de ARNr 16s. Resumen de datos. (2019) 27:104764. doi: 10.1016/j.dib.2019.104764
Resumen de PubMed | Texto completo de Crossref | Google Académico
93. Limón, G , Gamawa, AA , Ahmed, IA , Lyons, NA y Barba, PM . Características epidemiológicas e impacto económico de la dermatosis nodular contagiosa, la viruela ovina y la viruela caprina entre los agricultores de subsistencia del noreste de Nigeria. Front Vet Sci. (2020) 7. doi: 10.3389/fvets.2020.00008
Resumen de PubMed | Texto completo de Crossref | Google Académico
94. Greeff, JC , Paz, EA , Munyard, K , Schlink, AC , Smith, J , Karlsson, LJE, et al. Análisis del microbioma de la piel de ovejas resistentes o susceptibles al ataque de moscas de nalgas. Anim Prod Sci. (2021) 61:1774–80. doi: 10.1071/AN21063
95. Khalil, A , Batool, A y Arif, S . Microbioma sano del ganado y disbiosis en fenotipos enfermos. Rumiantes. (2022) 2:134–56. doi: 10.3390/rumiantes2010009
96. Clausen, M-L , Agner, T , Lilje, B , Edslev, SM , Johannesen, TB y Andersen, PS . Asociación de la gravedad de la enfermedad con el microbioma de la piel y las mutaciones del gen de la filagrina en la dermatitis atópica del adulto. JAMA Dermatol. (2018) 154:293–300. doi: 10.1001/jamadermatol.2017.5440
Resumen de PubMed | Texto completo de Crossref | Google Académico
97. Falentin, H , Rault, L , Nicolás, A , Bouchard, DS , Lassalas, J , Lamberton, P, et al. El análisis del microbioma de los pezones bovinos reveló una diversidad alfa reducida y cambios significativos en los perfiles taxonómicos en los barrios con antecedentes de mastitis. Microbiol frontal. (2016) 7:480. doi: 10.3389/fmicb.2016.00480
Resumen de PubMed | Texto completo de Crossref | Google Académico
98. Chu, Y , Yan, X , Gao, P , Zhao, P , Él, Y , Liu, J, et al. Detección molecular de una infección mixta por el virus de la viruela caprina, el virus Orf y Mycoplasma capricolum subsp. capripneumoniae en cabras. J Vet Diagn Invest. (2011) 23:786–9. doi: 10.1177/1040638711407883
Resumen de PubMed | Texto completo de Crossref | Google Académico
99. Sant’Ana, F. J. F.De , Leal, F. A. A. , Rabelo, R. E. y Vulcani, V. A. S ., Moreira, C. A. y Cargnelutti, J. F .,, et al. (2013). Coinfección por el virus vaccinia y un parapoxvirus similar al virus Orf en un brote de enfermedad vesicular en vacas lecheras en el centro-oeste de Brasil. J Vet Diagn Invest 25, 267–272. doi: 10.1177/1040638713475799
100. Bf, L . Fusobacterium necrophorum: sus características y papel como patógeno animal. Bacteriol Rev. (1977) 41:373–90. DOI: 10.1128/BR.41.2.373-390.1977
Resumen de PubMed | Texto completo de Crossref | Google Académico
101. Baracco, GJ . Infecciones causadas por estreptococos de los grupos C y G (Streptococcus dysgalactiae subsp. equisimilis y otros): aspectos epidemiológicos y clínicos. Microbiol Spectr. (2019) 7. DOI: 10.1128/microbiolspec. GPP3-0016-2018
Resumen de PubMed | Texto completo de Crossref | Google Académico
102. Lundberg, Å , Nyman, A , Unnerstad, HE y Waller, KP . Prevalencia de genotipos bacterianos y evolución de la mastitis clínica bovina por Streptococcus dysgalactiae y Streptococcus uberis. Acta Vet Scand. (2014) 56:80. doi: 10.1186/s13028-014-0080-0
Resumen de PubMed | Texto completo de Crossref | Google Académico
103. Casa, JA , Wilson, TM , el Nakashly, S , Karim, IA , Ismail, yo , el Danaf, N, et al. El aislamiento del virus de la dermatosis nodular contagiosa y del herpesvirus bovino-4 del ganado bovino en Egipto. J Vet Diagn Invest. (1990) 2:111–5. doi: 10.1177/104063879000200205
Resumen de PubMed | Texto completo de Crossref | Google Académico
104. Cunha, F , Jeon, SJ , Daetz, R , Vieira-Neto, A , Laporta, J , Jeong, KC, et al. Cuantificar patógenos uterinos conocidos y emergentes, y evaluar su asociación con metritis y fiebre en vacas lecheras. Teriogenología. (2018) 114:25–33. doi: 10.1016/j.theriogenology.2018.03.016
Resumen de PubMed | Texto completo de Crossref | Google Académico
105. Annandale, CH , Holm, DE , Ebersohn, K y Venter, EH . Transmisión seminal del virus de la dermatosis nodular contagiosa en novillas. Transbound Emerg Dis. (2014) 61:443–8. doi: 10.1111/tbed.12045
Resumen de PubMed | Texto completo de Crossref | Google Académico
106. Casal, J , Allepuz, A , Miteva, A , Pite, L , Tabakovsky, B , Terzievski, D, et al. Coste económico de los brotes de dermatosis nodular contagiosa en tres países de los Balcanes: Albania, Bulgaria y la Antigua República Yugoslava de Macedonia (2016-2017). Transbound Emerg Dis. (2018) 65:1680–8. doi: 10.1111/tbed.12926
Resumen de PubMed | Texto completo de Crossref | Google Académico
107. Rzewuska, M , Kwiecień, E , Chrobak-Chmiel, D , Kizerwetter-Świda, M , Stefańska, I y Gieryńska, M . Patogenicidad y virulencia de Trueperella pyogenes: una revisión. Int J Mol Sci. (2019) 20:2737. doi: 10.3390/ijms20112737
Resumen de PubMed | Texto completo de Crossref | Google Académico
Palabras clave: virus de la dermatosis nodular contagiosa, virus de la viruela ovina, metagenoma, comunidad bacteriana, secuenciación de escopeta, microbioma cutáneo, microbioma de lesiones cutáneas
Cita: Sharko FS, Mazloum A, Krotova AO, Byadovskaya OP, Prokhvatilova LB, Chvala IA, Zolotikov UE, Kozlova AD, Krylova AS, Grosfeld EV, Prokopenko AV, Korzhenkov AA, Patrushev MV, Namsaraev ZB, Sprygin AV y Toshchakov SV (2024) Perfil metagenómico de las comunidades virales y microbianas de las lesiones de la viruela del virus de la enfermedad cutánea nodular contagiosa y los huéspedes infectados por el virus de la viruela ovina. Frente. Vet. Sci. 11:1321202. doi: 10.3389/fvets.2024.1321202
Editado por:
Yasser Mahmmod, Escuelas Superiores de Tecnología, Emiratos Árabes Unidos
Revisado por:
Naveen Kumar, ICAR-Instituto Nacional de Enfermedades Animales de Alta Seguridad (ICAR-NIHSAD), India
Meng Zhang, Universidad Agrícola de Mongolia Interior, China
Mohamed Elsokary, Universidad de Benha, Egipto
Derechos de autor © 2024 Sharko, Mazloum, Krotova, Byadovskaya, Prokhvatilova, Chvala, Zolotikov, Kozlova, Krylova, Grosfeld, Prokopenko, Korzhenkov, Patrushev, Namsaraev, Sprygin y Toshchakov. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia Creative Commons Attribution License (CC BY).
*Correspondencia: Stepan V. Toshchakov, stepan.toshchakov@gmail.com
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o afirmación que pueda hacer su fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía