
Salmonella enterica induce cambios específicos en el microbioma intestinal de los cerdos

Salmonella enterica induce cambios específicos de la biogeografía en el microbioma intestinal de los cerdos
 Joao Carlos Gomes-Neto1,2†
Joao Carlos Gomes-Neto1,2†  Natasha Pavlovikj3†
Natasha Pavlovikj3†  Nate Korth1,2†
Nate Korth1,2†  Samantha A. Naberhaus4,5 Bailey Arruda5
Samantha A. Naberhaus4,5 Bailey Arruda5  Andrew K. Benson1,2
Andrew K. Benson1,2  Amanda J. Kreuder4,5*
Amanda J. Kreuder4,5*- 1Departamento de Ciencia y Tecnología de los Alimentos, Universidad de Nebraska-Lincoln, Lincoln, NE, Estados Unidos
- 2Nebraska Food for Health Center, University of Nebraska-Lincoln, Lincoln, NE, United States
- 3Holland Computing Center, Universidad de Nebraska-Lincoln, Lincoln, NE, Estados Unidos
- 4Departamento de Microbiología Veterinaria y Medicina Preventiva, Iowa State University, Ames, IA, Estados Unidos
- 5Department of Veterinary Diagnostic and Production Animal Medicine, College of Veterinary Medicine, Iowa State University, Ames, IA, Estados Unidos
Los cerdos son un reservorio importante de una serie de serovares zoonóticos de linaje I de Salmonella enterica que incluyen Derby, Typhimurium y 4,[5],12:i:- (también conocido como Typhimurium monofásico). En este estudio, evaluamos la composición del microbioma gastrointestinal (GI) de cerdos en diferentes compartimentos intestinales y las heces después de la infección con serovariedades zoonóticas específicas de S. enterica (S. Derby, S. Monofásico, y S. typhimurium). Se realizó un análisis del microbioma basado en ARNr 16S para evaluar los cambios en el microbioma GI en términos de diversidad (alfa y beta), estructura y volatilidad de la comunidad, y alteraciones específicas de taxones en la biogeografía GI (intestino delgado y grueso, heces) y días posteriores a la infección (DPI) 2, 4 y 28; Estos resultados se compararon con fenotipos de enfermedad medidos como cambios histopatológicos. Como se informó anteriormente, solo S. Monofásico y S. Typhimurium indujeron alteraciones morfológicas que marcaron un medio inflamatorio restringido al intestino grueso en este modelo experimental. S. Typhimurium solo indujo cambios significativos en los niveles de diversidad alfa (índices de Simpson y Shannon) y beta, específicamente en el pico de inflamación en el intestino grueso y las heces. También se observó un aumento de la dispersión de la comunidad y volatilidad en el ápice colónico y los microbiomas fecales para S. Typhimurium. Los tres serovares de Salmonella alteraron la estructura de la comunidad medida por redes de co-ocurrencia; esto fue más prominente en DPI 2 y 4 en muestras de ápice colónico. A nivel taxonómico del género, una amplia gama de bacterias productoras de ácidos grasos de cadena corta (AGCC) se alteraron y, a menudo, disminuyeron durante el pico de inflamación en DPI 2 y 4 dentro del ápice colónico y muestras fecales. Entre todas las bacterias productoras de AGCC putativas, Prevotella mostró un amplio patrón de correlación negativa con las puntuaciones de la enfermedad en el pico de inflamación. Además, se encontró que Prevotella 9 se redujo significativamente en todos los grupos infectados por Salmonella en comparación con el control en DPI 4 en el ápice colónico. En conclusión, este trabajo aclara además que los distintos serovares zoonóticos relacionados con cerdos de S. enterica pueden inducir alteraciones compartidas (alta resiliencia) y únicas (resistencia alterada) en la biogeografía del microbioma intestinal, lo que ayuda a informar futuras investigaciones de modificaciones dietéticas destinadas a aumentar la resistencia a la colonización contra Salmonella a través de alteraciones del microbioma gastrointestinal
1. Introducción
Según los Centros para el Control y la Prevención de Enfermedades (CDC), aproximadamente 50 millones de personas adquieren enfermedades asociadas a los alimentos en los Estados Unidos anualmente (1). La salmonela es un patógeno global, con salmonelosis transmitida por los alimentos que conduce a más de un millón de infecciones, miles de hospitalizaciones y cientos de muertes anuales solo en los Estados Unidos (2). Los alimentos y productos relacionados con el ganado son la fuente zoonótica predominante de salmonelosis humana (3). La salmonela se divide principalmente en dos especies, a saber, S. enterica y S. bongori, con la gran mayoría de los serovares zoonóticos y aislados clínicos humanos pertenecientes al linaje I de S. enterica subsp. enterica (denominado linaje I de S. enterica) (4). Los cerdos son un reservorio importante de una serie de serovares del linaje zoonótico de S. enterica I, incluidos Derby, 4,[5],12:i:- (también conocido como Typhimurium monofásico), y Typhimurium (3, 5-7). Los tres serovares son capaces de residir en el tracto gastrointestinal (GI) de los cerdos y pueden propagarse a través de la cadena alimentaria y causar brotes humanos (3, 5, 6, 8). Salmonella Derby se aisló inicialmente de los seres humanos tras el consumo de pasteles de cerdo contaminados (9), y ahora se espera que se encuentre en todos los principales centros de producción porcina en todo el mundo (5, 6); también se ha asociado recientemente con brotes infantiles en China (10). Por otro lado, la población de S. Typhimurium se puede estratificar en dos sublinajes divergentes: Typhimurium y 4,[5],12:i:- (monofásico) (11, 12). Salmonella Los aislados de Typhimurium se conocen típicamente como huéspedes generalistas y pueden residir en el tracto gastrointestinal del ganado, aves de corral y cerdos, todos los cuales pueden servir como fuente de brotes humanos (3, 13), mientras que S. Monofásico es un pathovar zoonótico emergente que puede aislarse del ganado vacuno y las aves de corral, pero se encuentra más comúnmente en el tracto gastrointestinal de los cerdos (8, 13, 14). S. monofásico deriva de S. Typhimurium pero contiene únicamente un elemento integrador y conjugativo (ICE) llamado isla genómica de Salmonella (SGI)-3/4 que confiere fenotípicamente resistencia a metales pesados como cobre, arseniato y plata y se inserta en un lugar que interrumpe la variación de fase normal (11, 15-18). Fenotipos de resistencia al cobre de S. Monofásico bajo condiciones aeróbicas y anaeróbicas sugieren que SGI-3/4 puede contribuir a la supervivencia en entornos naturales (por ejemplo, depósitos de agua e instalaciones de alimentos) y puede conducir preferentemente a una ventaja de aptitud en cerdos potencialmente debido a la utilización de cobre dietético (11, 19). Además, ha habido un número creciente de informes que describen la diseminación mundial de S multirresistente (MDR). Aislamientos monofásicos, que representan una amenaza adicional para la salud pública (20–24).
Con el tiempo, se han hecho muchos intentos para mitigar la Salmonella (centrada en gran medida en los últimos años en S. typhimurium) en la industria porcina mediante el desarrollo de estrategias basadas en granjas para disminuir la prevalencia (por ejemplo, cambios en la dieta y vacunación), mejorando el diagnóstico y la vigilancia, y aumentando las prácticas y regulaciones de higiene en las instalaciones de producción de alimentos (3, 7, 25–30). Se han implementado modificaciones dietéticas que incluyen el uso de antimicrobianos, probióticos y prebióticos para aumentar la resistencia a la colonización contra Salmonella, parcialmente a través de alteraciones del microbioma gastrointestinal (25, 31, 32). Además, se ha demostrado recientemente que la composición del microbioma GI está asociada con la variación en el transporte de ambas S. Monofásico y S. typhimurium (32–34), encontrándose que Prevotella sp. se correlaciona negativamente con el transporte y el desprendimiento (33–35). En el estado estacionario, se espera que el tracto gastrointestinal porcino tenga una composición microbiana central única en distintos sitios anatómicos (36), con Prevotella como taxón clave en cerdos destetados hasta las etapas de finalización (36, 37); esto sugiere que las firmas basadas en microbiomas de resistencia a la colonización a Salmonella también son probablemente distintas en diferentes sitios anatómicos debido a la variación del tropismo tisular. Considerando S. La infección e inflamación monofásica y por S. typhimurium a menudo se puede compartimentar en segmentos específicos del intestino grueso (por ejemplo, tejidos colónicos ciego y ápice) (8), se necesitan más estudios para evaluar la biogeografía del microbioma gastrointestinal tras la infección de cerdos con distintos serovares zoonóticos de S. enterica para aprovechar las firmas predecibles de resistencia a la colonización prácticamente desplegables en los sistemas modernos de producción porcina. También es importante tener en cuenta que S. typhimurium, en general, puede explotar el medio inflamatorio del huésped para obtener acceso epitelial transintestinal y persistir en los cerdos (32, 38). La resistencia a la colonización a Salmonella es un rasgo complejo que implica capacidades de resistencia (capacidad de resistir una perturbación) y resiliencia (capacidad de recuperarse de una perturbación) de la microbiota gastrointestinal (39).
Por lo tanto, en este estudio exploramos más a fondo un modelo experimental de infección previamente publicado en cerdos para evaluar cómo los aislados específicos de serovares zoonóticos conocidos de S. enterica (Derby, Monofásico y Typhimurium) alteran la biogeografía del microbioma GI horas extras (día después de la infección – DPI). Específicamente, realizamos la secuenciación del amplicón 16S rRNA para mapear taxones bacterianos que podrían diferenciarse entre grupos infectados e inoculados simulados en muestras del intestino pequeño y grueso, así como en heces, mientras medimos y analizamos: (1) diversidad alfa y beta; (2) estructura y volatilidad de la comunidad; (3) abundancia relativa entre taxones; y (4) correlación entre taxones específicos (abundancia relativa) y puntuaciones histopatológicas. En general, nuestros resultados mostraron (1) solo S. Monofásico y Typhimurium podrían provocar inflamación transitoria manifiesta (DPI 2 y 4) en el intestino grueso junto con perturbaciones del microbioma gastrointestinal; (2) las diferencias de diversidad alfa y beta fueron más claras para los grupos monofásico y Typhimurium (firma única) en DPI 2 y 4 en el ápice del colon espiral y el contenido fecal; y (3) a nivel taxonómico se indujeron cambios únicos y compartidos de serovar, demostrando en promedio un cuello de botella inflamatorio transitorio (baja resistencia) que impactó directamente la proporción y las correlaciones fenotípicas con las bacterias productoras de ácidos grasos de cadena corta (AGCC).
2. Materiales y métodos
2.1. Estudios en animales
Una descripción completa de los estudios en animales de los que se tomaron las muestras utilizadas en este estudio se proporciona en Naberhaus et al. (8). Si bien se recolectaron muestras metagenómicas y se generaron datos de secuenciación para los tres estudios experimentales descritos anteriormente, solo se utilizaron datos del ensayo 2 para este análisis, ya que proporcionaron un análisis longitudinal y biogeográfico de múltiples serovariedades de Salmonella en múltiples animales. En este estudio, todos los cerdos (sexo mixto) tenían 5 semanas de edad al comienzo del ensayo; esta edad se eligió en base a los datos epidemiológicos retrospectivos acumulados en la última década por el Laboratorio de Diagnóstico Veterinario (VDL) de la Universidad Estatal de Iowa (ISU). Mediante pruebas PCR agrupadas e individuales en muestras fecales, todos los cerdos dieron negativo para Salmonella antes de la inoculación; todos los cerdos también dieron negativo para el virus del síndrome reproductivo y respiratorio porcino (PRRSV) y el virus de la diarrea epidémica porcina (PEDV) antes del inicio del estudio. Los cerdos fueron identificados individualmente y asignados a los siguientes tratamientos: (1) 12 cerdos inoculados simuladamente con caldo estéril Mueller Hinton (MH) sirvieron como control negativo; (2) 20 cerdos recibieron inoculación oral con S. Hongo; (3) 20 cerdos recibieron inoculación oral con S. 4,[5],12:i:- (S. monofásica); y (4) 20 cerdos recibieron inoculación oral con S. Typhimurium. Para evitar la contaminación cruzada, tras la asignación del tratamiento, cada grupo se alojó por separado (cuatro cerdos por corral en todos los tratamientos) durante la duración del estudio. Las inoculaciones se realizaron utilizando una combinación de 8 ml de sonda oral y 2 ml de hisopado directamente en la parte posterior de la boca asegurando la exposición de las amígdalas para un total de 10 ml de 1 × 108 UFC/ml Salmonella. Los cerdos fueron alimentados ad libitum excepto durante 12 h antes de la inoculación y fueron sacrificados usando sobredosis de barbitúricos. Todos los estudios con animales fueron aprobados por el Comité Institucional de Cuidado y Uso de Animales de la Universidad Estatal de Iowa (IACUC) antes del inicio (11-16-8,391-S).
2.2. Recogida de muestras
Antes de cualquier otra recolección de muestras (es decir, temperatura, muestra fecal para cultivo), se recolectaron hisopos fecales para el análisis del microbioma basado en ARNr 16S del recto de todos los cerdos en DPI 0 y 2, y todos los cerdos vivos en DPI 4, 7, 14, 21 y 28. Estas muestras se arremolinaron en solución salina tamponada con fosfato estéril de Dubelco (1X DPBS, Corning, Ref 21-030-CM) e inmediatamente se congelaron en nitrógeno líquido después de la recolección y se mantuvieron a -80 ° C hasta el procesamiento de la muestra. El contenido del tracto gastrointestinal se recogió del intestino delgado (íleon) y del intestino grueso (ápice del colon espiral) en el momento de la necropsia (DPI 2, 4 o 28). En DPI 2 y 4, se seleccionaron cinco cerdos por grupo infectado con Salmonella y tres cerdos de control para eutanasia y necropsia en función de la gravedad de los signos clínicos (es decir, cerdos infectados con Salmonella que demostraron los signos clínicos más graves basados en una combinación de temperatura rectal y puntuación fecal). A los cerdos restantes después de DPI 4 (10 por grupo experimental; 6 en el grupo de control) se les permitió completar el estudio y fueron sacrificados para la recolección de muestras en DPI 28. En el momento de la eutanasia, se realizó una necropsia para evaluar las lesiones macroscópicas. Se recolectaron muestras de tejido y se colocaron en formalina al 10% para la evaluación histopatológica ciega del yeyuno, el íleon, el ciego, el colon espiral medio, el ápice del colon espiral (ápice colónico) y el recto. Los contenidos del ápice ileal y colónico recolectados en el momento de la necropsia se congelaron rápidamente en nitrógeno líquido y se mantuvieron a -80 ° C hasta el procesamiento de la muestra. También se tomaron raspados de la mucosa ileal y el ápice de la mucosa espiral del colon, congelados en nitrógeno líquido y almacenados a -80 ° C.
2.3. Sistema de puntuación histopatológica
Una descripción detallada del sistema de puntuación histopatológica para cada sitio examinado se puede encontrar en Naberhaus et al. (8). En resumen, las puntuaciones histológicas se asignaron en función de la presencia de neutrófilos, ulceración y profundidad de la cripta (solo intestino grueso); Se asignaron puntos adicionales si había inflamación submucosa o abscesos de cripta. Con el fin de correlacionar la enfermedad (es decir, la puntuación histológica) con los datos metagenómicos, se desarrolló una puntuación histopatológica acumulativa que combina los datos registrados para los cuatro sitios del intestino grueso (ciego, colon espiral medio, ápice del colon espiral y recto) y los sitios del intestino delgado (yeyuno proximal, medio y distal; íleon). Cabe destacar que la puntuación histopatológica del ápice colónico también se utilizó para verificar asociaciones con taxones de interés enriquecidos en el contenido del ápice.
2.4. Preparación y secuenciación del ADN
La extracción de ADN se realizó utilizando el kit de ARN/ADN patógeno MagMAX™ (Thermo Fisher Scientific, Waltham, MA, Estados Unidos) y un instrumento Kingfisher Flex (Thermo Fisher Scientific) siguiendo las instrucciones del fabricante. Las muestras se enviaron a la instalación de ADN de la Universidad Estatal de Iowa para la preparación de la biblioteca y la secuenciación de la metagenómica 16S. El ADN total extraído se amplificó utilizando cebadores (515F y 806R) específicos para las regiones V4 del gen 16S rRNA bacteriano y la preparación de la biblioteca se realizó como se describió anteriormente (40-43). Las siguientes muestras de preparación de la biblioteca se ejecutaron en 150 ciclos de extremo pareado en un instrumento Illumina MiSeq. Se enviaron y secuenciaron un total de 721 muestras en tres carriles con un número máximo de muestras por carril de 240. Se incluyeron controles negativos consistentes en hisopos estériles en DPBS, así como un control de extracción negativo; controles positivos consistentes en estándares de la Comunidad Microbiana de Zymobiomics (catálogo #D6305 y D6300; Zymo Research Corporation, Irvine, CA) también se incluyeron en todas las placas para garantizar la consistencia entre las placas en la secuenciación.
2.5. Análisis de datos de ARNr 16S
Los datos multiplexados generados a partir de los tres carriles MiSeq se analizaron utilizando QIIME2 (44), versión 2021.11. Las lecturas de extremo pareado demultiplexadas de cada carril se eliminaron y las lecturas con una puntuación de calidad por debajo del valor predeterminado de Q20 se recortaron y filtraron con DADA2 (45). Para acelerar los análisis computacionales, este paso se realizó por separado en cada carrera de los tres carriles. A continuación, la tabla y las secuencias de las ejecuciones separadas de DADA2 se fusionaron con qiime feature-table merge y qiime feature-table merge-seqs. Luego, los datos se filtraron para incluir solo muestras recolectadas del ensayo 2. La tabla de características actualizada se utilizó más tarde para elegir una profundidad de muestreo para los análisis de diversidad. Los análisis taxonómicos y la asignación de unidades taxonómicas a cada una de las secuencias representativas se realizaron con el clasificador de características qiime classify-sklearn y la base de datos de referencia SILVA (silva-138.1-ssu-nr99). Las mitocondrias y la secuencia de cloroplastos se eliminaron aún más utilizando la tabla de filtros de taxones qiime. Finalmente, se realizó un árbol filogenético, así como diversas métricas de diversidad (alfa y beta) y análisis de similitud utilizando QIIME2.
2.6. Análisis estadístico
Los resultados taxonómicos de QIIME2 se procesaron para el control de calidad hasta el modelado estadístico utilizando R versión 4.0.5. La biblioteca tidyverse (versión 1.3.1) se utilizó para la exploración, el análisis y el trazado de datos. Todas las muestras mantenidas en el análisis pasaron los siguientes criterios combinados: «porcentaje de entrada pasada filtro» > 75 y «porcentaje de entrada fusionada» > 60 y «porcentaje de entrada no quimérica» > 60. A partir de entonces, solo se incluyeron en el análisis los taxones clasificados como «Bacterias» y que tenían información a nivel de género. Ni los taxones clasificados como «Archaea» ni los clasificados como no cultivados a nivel de género fueron incluidos en el análisis estadístico de la composición de la microbiota. Se calcularon proporciones individuales basadas en taxones por animal teniendo en cuenta el DPI, el tratamiento y el tipo de muestra. Se utilizó un análisis unidireccional de varianza (ANOVA) para evaluar el efecto del tratamiento sobre las puntuaciones histopatológicas, las métricas de diversidad alfa (D de Shannon y Simpson), la descomposición de la diversidad beta y el análisis de volatilidad (PC1 o PC2) y la abundancia relativa basada en taxones individuales (proporción). Todos los modelos ANOVA se realizaron utilizando la función aov() en R. Si el efecto del tratamiento fue significativo (p < 0,05) en el modelo ANOVA, se utilizó una prueba T por pares para evaluar la diferencia entre los grupos (p < 0,05), a menos que se indique lo contrario. Todas las pruebas T por pares se realizaron utilizando la función pairwise.t.test() en R sin un ajuste del valor p y utilizando una desviación estándar agrupada por defecto. Los índices D de diversidad alfa de Shannon y Simpson se calcularon con la función diversity() en R de la biblioteca vegana (versión 2.6.2). La diversidad beta se calculó utilizando la función vegdist (método = «bray», binario = «FALSO», na.rm. = «VERDADERO») de la biblioteca vegana (versión 2.6.2) mientras se usaba una matriz de distancia de Bray-Curtis de datos no binarios y se eliminaban todos los valores faltantes del análisis. Para el análisis de coordenadas principales (PCoA) se utilizó un modelo de escala multidimensional clásico para reducir los datos (matriz de distancia de Bray-Curtis) a dos dimensiones (2 coordenadas principales – PC), utilizando la función cmdscale (k = 2) en R de la biblioteca de estadísticas (versión 4.0.5). Se utilizó un PERMANOVA y análisis de similitud (ANOSIM) para calcular el efecto del tratamiento de la diversidad beta (distancias Bray-Curtis), utilizando las funciones adonis2(permutaciones = 999, método = «bray») y anosimo (método = «bray») en R, respectivamente, de la biblioteca vegana (versión 2.6.2). Correlaciones de Pearson entre logaritmostáticos2 La proporción de taxones transformados (tanto del ápice colónico como de las heces) y las puntuaciones histopatológicas del ápice intestinal/colónico grande se calcularon utilizando la función cor_test() en R utilizando parámetros predeterminados, como parte de la biblioteca Rstatix (versión 0.7.2). El análisis LEfSe para la identificación de taxones diferenciadores se realizó con la plataforma Galaxy con un ajuste del valor p de Wilcoxon a 0.1, debido a los pequeños tamaños de muestra en todos los tratamientos, y al eliminar el taxón o las características con recuentos cero en todas las muestras (46, 47). En última instancia, los taxones más diferenciadores se seleccionaron en función de (1) la proporción relativa general superior al 2%; (2) Resultados LEfSe; y (3) taxones centrales sobre análisis de redes de co-ocurrencia. Además, se crearon patrones de co-ocurrencia individualizados o promedio (composición de la comunidad) de los cambios del microbioma utilizando taxones en diferentes niveles de resolución (por ejemplo, familia, género) o los taxones más diferenciadores, respectivamente. Los patrones basados en promedio se crearon utilizando un registro2 transformación de la proporción media para cada taxón entre tratamientos y DPI. En todos los taxones principales identificados, las proporciones se calcularon por animal / muestra individual, pero para cada animal / muestra, las proporciones se sumaron para eliminar la redundancia taxonómica y la inexactitud al hacer los cálculos finales (por ejemplo, todos los taxones que contienen una «g__Prevotella» en el nombre se combinaron con Prevotella para los cálculos a nivel de género). Para todas las funciones de R utilizadas, si no se indicaba, se utilizaban los parámetros predeterminados para los cálculos.
2.7. Análisis de redes de coocurrencia
El análisis de red y la visualización de datos se realizaron utilizando el paquete NetCoMi dentro del marco estadístico R (v4.2.2) (48). Cada red se construyó para cada tratamiento, empleando una transformación logarítmica centrada y correlaciones basadas en Spearman entre los 50 taxones microbianos más abundantes. La interpretación de los datos se resumió en función de que cada nodo representa un taxón bacteriano, y el tamaño de cada nodo se escala de acuerdo con su centralidad. Las aristas representaban asociaciones entre taxones, con asociaciones positivas coloreadas en verde y negativas en rojo; El grosor de cada borde corresponde a la fuerza de la asociación. No se mostraron las aristas que representaban un valor inferior a 0,5. Los taxones fueron coloreados en base a grupos calculados en la construcción de la red.
2.8. Plataformas computacionales
Todos los análisis bioinformáticos del microbioma 16S rRNA se realizaron en Crane, uno de los clústeres de computación de alto rendimiento de Linux en el Centro de Computación Holland (HCC) de la Universidad de Nebraska.1
3. Resultados
3.1. Visión general del enfoque experimental y el fenotipo de la enfermedad en serovares
Utilizando muestras recogidas de nuestro trabajo anterior evaluando la patogenicidad de S. Derby, S. Monofásico, y S. Typhimurium (8) en cerdos, se realizó un análisis de microbioma basado en ARNr 16S para evaluar el impacto de diferentes serovares zoonóticos asociados a cerdos del linaje I de S. enterica en la biogeografía del microbioma GI. La Figura 1A ilustra el diseño experimental y los objetivos del estudio. Las cifras suplementarias S1A-C muestran el número de animales analizados por tratamiento y DPI después de la curación de datos del microbioma. Como se demuestra en la Figura 1B, solo S. Monofásico y S. Typhimurium indujeron inflamación manifiesta significativa, restringida al intestino grueso, durante DPI 2 y 4 después de la infección (p < 0,05); no se observaron lesiones inflamatorias con la infección por S. Derby o en el intestino delgado para S. Monofásico y S. Typhimurium, como se estableció anteriormente (8). No se encontraron diferencias significativas en las puntuaciones histopatológicas entre S. Monofásico y S. Typhimurium en el intestino grueso (p ≥ 0,05). Cuando se examinó por separado la histopatología del ápice colónico, no se encontraron diferencias significativas debido al pequeño tamaño y variabilidad de la muestra, aunque en promedio S. Los animales monofásicos e infectados por S. typhimurium presentaron puntuaciones más altas (Figura suplementaria S2). Según nuestro informe anterior (8), la eliminación de Salmonella en este estudio alcanzó un máximo de DPI 2-4 simultáneamente con la inflamación máxima y, por lo general, disminuyó de 7 a 10 DPI. En general, ambos S. Monofásico y S. Typhimurium tenían una cinética de enfermedad comparable restringida al intestino grueso.
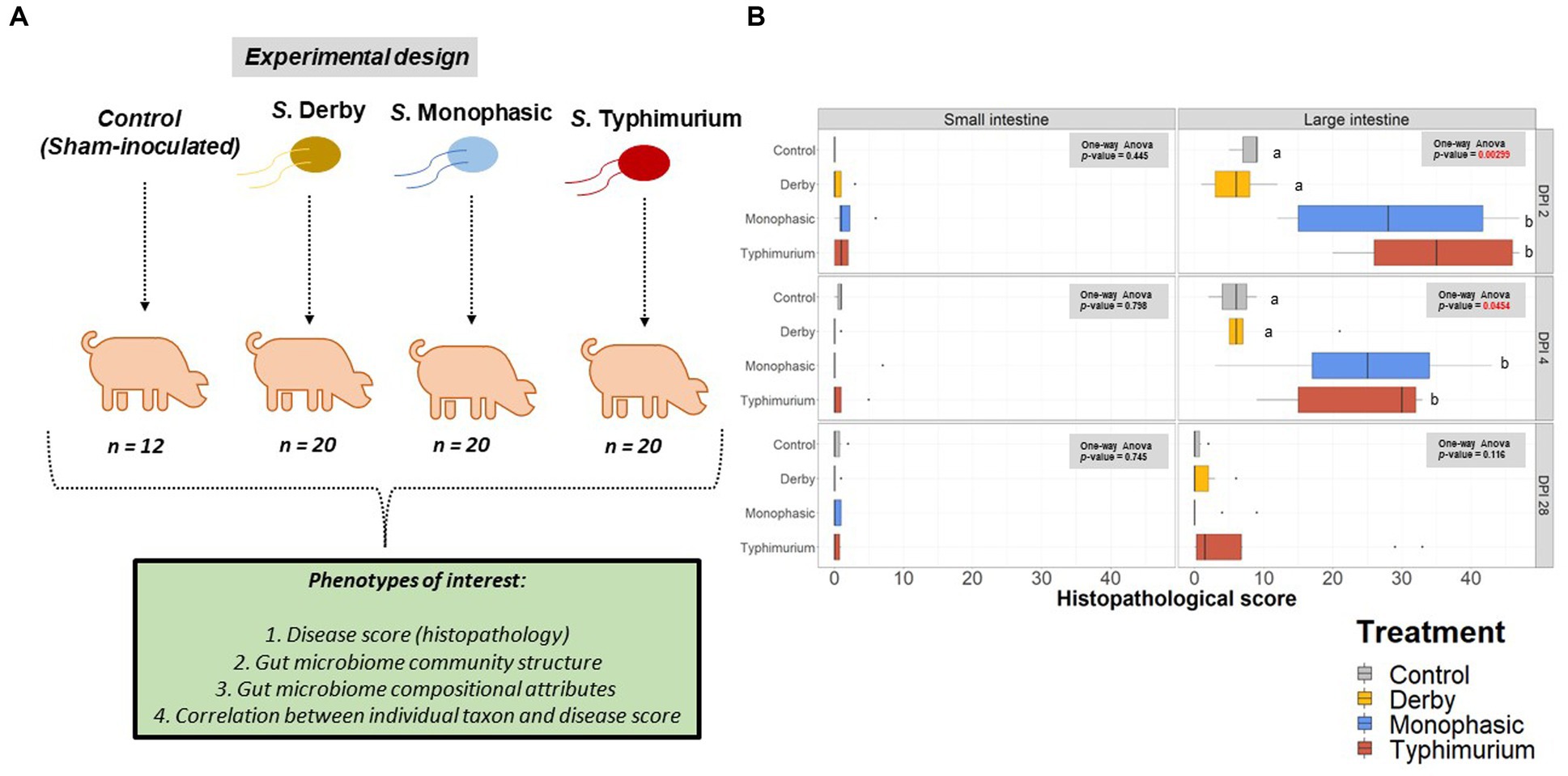 Figura 1. Diseño experimental y puntuaciones histopatológicas. (A) Flujo de trabajo de diseño experimental que incluye el número de animales por tratamiento y fenotipos de interés, incluida la histopatología (puntuación de la enfermedad), la composición del microbioma GI, las abundancias relativas de taxones y la correlación entre las puntuaciones individuales de taxones e histopatologías. (B) Puntuaciones histopatológicas a través del intestino delgado (íleon) y el intestino grueso en DPI 2, 4 y 28. Se utilizó un ANOVA unidireccional para medir el efecto del tratamiento (grupos control vs. grupos infectados por Salmonella) (p < 0,05). Cuando el efecto del tratamiento fue significativo basado en el análisis ANOVA unidireccional (valores de p marcados en rojo), las comparaciones por pares se realizaron mediante una prueba T bilateral (p < 0,05). Diferentes letras en superíndice indican diferencias significativas entre los tratamientos. El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
Figura 1. Diseño experimental y puntuaciones histopatológicas. (A) Flujo de trabajo de diseño experimental que incluye el número de animales por tratamiento y fenotipos de interés, incluida la histopatología (puntuación de la enfermedad), la composición del microbioma GI, las abundancias relativas de taxones y la correlación entre las puntuaciones individuales de taxones e histopatologías. (B) Puntuaciones histopatológicas a través del intestino delgado (íleon) y el intestino grueso en DPI 2, 4 y 28. Se utilizó un ANOVA unidireccional para medir el efecto del tratamiento (grupos control vs. grupos infectados por Salmonella) (p < 0,05). Cuando el efecto del tratamiento fue significativo basado en el análisis ANOVA unidireccional (valores de p marcados en rojo), las comparaciones por pares se realizaron mediante una prueba T bilateral (p < 0,05). Diferentes letras en superíndice indican diferencias significativas entre los tratamientos. El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
3.2. Resumen de los resultados de la secuenciación del amplicón del ARNr 16S
El primer carril MiSeq produjo 15,135,350 lecturas de extremo pareado, el segundo 15,825,865 lecturas de extremo pareado y el tercero 11,631,035 lecturas de extremo pareado, donde las lecturas hacia adelante y hacia atrás y los códigos de barras respectivos se almacenaron en archivos separados. Después de filtrar, eliminar ruido y fusionar, el número de lecturas resultantes fue de 7,983,113, 8,284,340 y 5,884,840 para los carriles 1, 2 y 3, respectivamente. Después de fusionar las ejecuciones individuales y filtrar los datos para incluir solo muestras del ensayo 2, el número total de muestras fue de 629 y el número total de secuencias representativas (características o ASV) fue de 5.774, con una frecuencia total de 18.738.198. La longitud mínima de las características (secuencias) fue de 152 pb, y la longitud máxima fue de 278 pb, siendo 252,32 la longitud media. Después de eliminar las secuencias de mitocondrias y cloroplastos, el número total de muestras finales utilizadas en nuestros análisis fue de 626 y el número total de secuencias representativas (características o ASV) fue de 5.733, con una frecuencia total de 18.729.244. La longitud mínima de las características (secuencias) fue de 152 pb, y la longitud máxima fue de 255 pb, siendo 252,38 la longitud media. Debido a la mala calidad de la secuencia (ver sección 2.6), no se pudo realizar el análisis de los raspados ileal y del ápice colónico. Para comparar la diversidad a través del contenido ileal, del ápice colónico y fecal en el mismo punto de tiempo para cada animal, el análisis se centró principalmente en muestras recolectadas en DPI 2, 4 y 28, excepto un análisis temporal transversal para muestras fecales en DPI 0, 2, 4, 7, 14, 21 y 28.
3.3. Los cambios en la diversidad alfa y beta se captan de forma aguda en el lugar de la inflamación y las heces
Las diferencias biogeográficas en la diversidad alfa se analizaron utilizando los índices D y Shannon de Simpson a través del ileal, el ápice colónico y el contenido fecal para DPI 2, 4 y 28. Tanto los índices de Simpson como los de Shannon destacaron que, en promedio, solo la infección por S. typhimurium redujo significativamente la diversidad alfa en muestras fecales en DPI 2 y en el intestino grueso (ápice colónico) en DPI 4 (pico de inflamación) (Figuras 2A, B) (p < 0.05). No obstante, los cambios en la diversidad alfa fueron transitorios ya que no hubo efecto del tratamiento en DPI 28 para ambos índices (p ≥ 0,05). Cabe destacar que, en promedio, la diversidad alfa del grupo infectado por S. typhimurium se restablece en muestras de ápice colónico por DPI 28 (Figuras 2A, B). Un análisis transversal más secuencial de la diversidad alfa en el contenido fecal también reveló una disminución transitoria en el grupo infectado por S. typhimurium por DPI 21 (p < 0,05) (Figuras suplementarias S3, S4). Curiosamente, los índices D de Shannon y Simpson sugieren que después del pico de inflamación (DPI 4), ambos S. Los grupos monofásicos y S. typhimurium tendieron a permanecer en promedio más bajos en diversidad en comparación con DPI 0 en muestras fecales (Figuras suplementarias S3, S4). A nivel de diversidad beta (distancia de Bray-Curtis), la dispersión de la comunidad reflejó los resultados de la diversidad alfa con cambios significativos capturados en DPI 4 para el ápice colónico y el contenido fecal (Figura 3) (p < 0,05). Cabe destacar que solo se encontraron cambios significativos en la diversidad beta en DPI 2 para el contenido fecal (p < 0,05), mientras que no se encontraron cambios significativos en el contenido ileal. Sin embargo, es importante destacar que la dispersión de la comunidad se observó más claramente entre los tratamientos en el pico de inflamación en el ápice colónico (valor de p = 0,007 y R-cuadrado = 0,33), basado en los resultados de PERMANOVA (Figura 3 – DPI 4 para muestras de ápice colónico). Un valor R-cuadrado más alto refleja un efecto biológico más fuerte del tratamiento, ya que explica más de la variabilidad total en los datos.
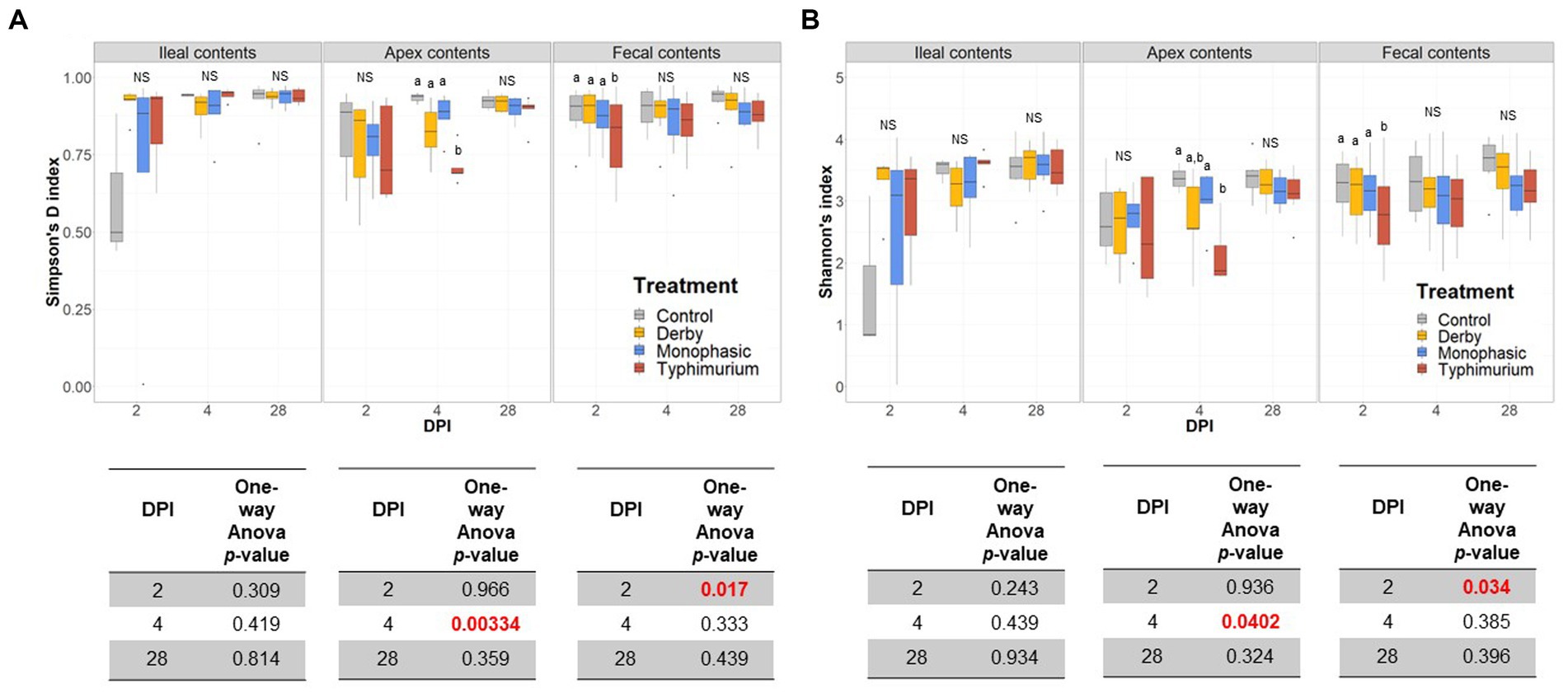 Figura 2. Análisis de diversidad alfa de la composición del microbioma GI en grupos de control vs. infectados por Salmonella. (A,B) Índices D y Shannon de Simpson de diversidad alfa en tratamientos y tipos de muestras, incluidos los contenidos ileal, del ápice colónico y fecal de cerdos en DPI 2, 4 y 28, respectivamente. Para ambas métricas de diversidad alfa, se utilizó un análisis ANOVA unidireccional para medir el efecto del tratamiento (p < 0,05). Cuando el efecto del tratamiento fue significativo según el análisis ANOVA unidireccional (valores de p marcados en rojo), las comparaciones por pares se realizaron mediante una prueba T bilateral (p < 0,05). Diferentes letras en superíndice indican diferencias significativas entre los tratamientos. El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
Figura 2. Análisis de diversidad alfa de la composición del microbioma GI en grupos de control vs. infectados por Salmonella. (A,B) Índices D y Shannon de Simpson de diversidad alfa en tratamientos y tipos de muestras, incluidos los contenidos ileal, del ápice colónico y fecal de cerdos en DPI 2, 4 y 28, respectivamente. Para ambas métricas de diversidad alfa, se utilizó un análisis ANOVA unidireccional para medir el efecto del tratamiento (p < 0,05). Cuando el efecto del tratamiento fue significativo según el análisis ANOVA unidireccional (valores de p marcados en rojo), las comparaciones por pares se realizaron mediante una prueba T bilateral (p < 0,05). Diferentes letras en superíndice indican diferencias significativas entre los tratamientos. El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
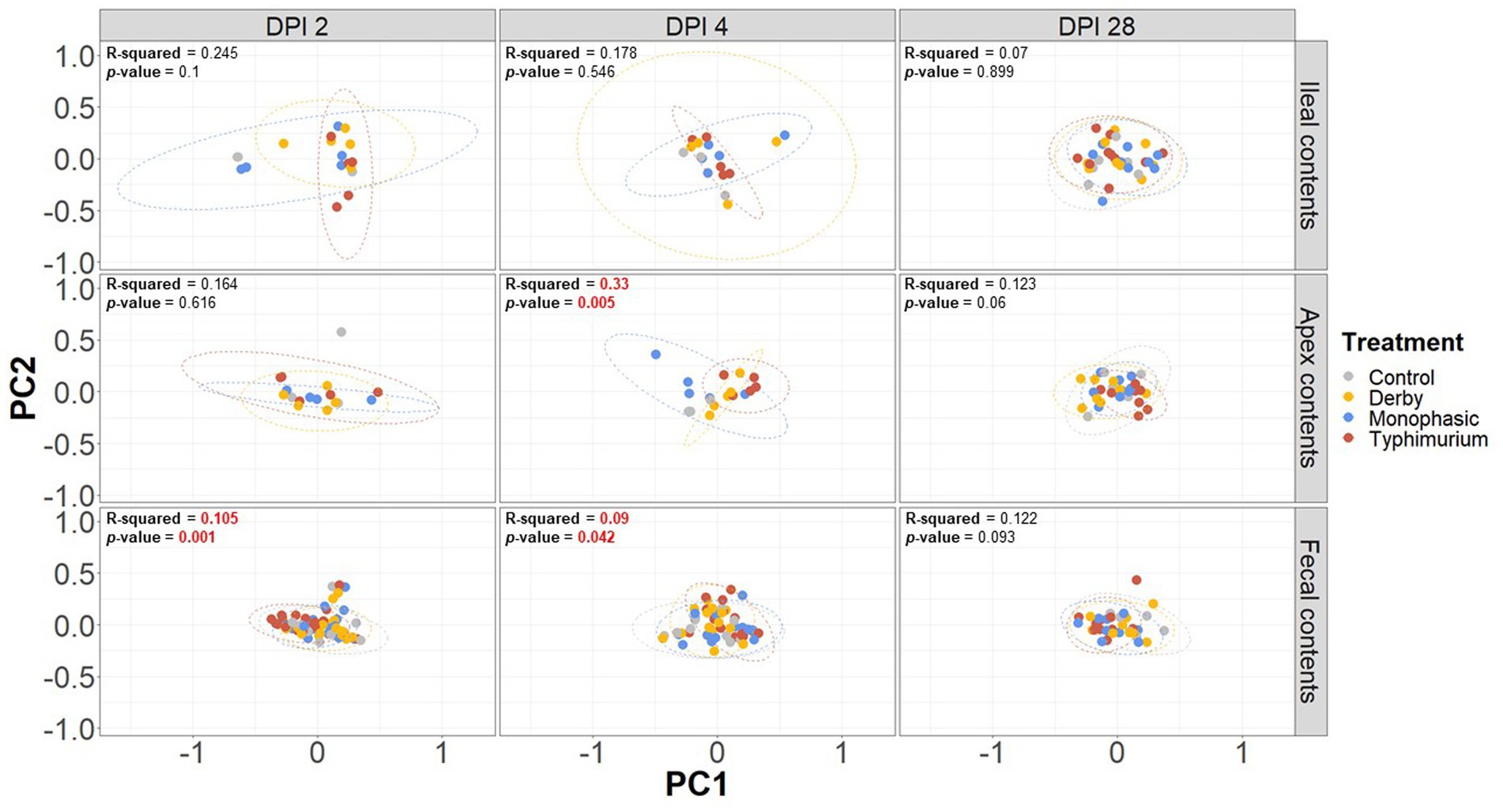 Figura 3. Análisis de la diversidad beta de la composición del microbioma GI en grupos de control frente a grupos infectados por Salmonella. Se utilizó la matriz de distancia de Bray-Curtis para calcular la diversidad beta entre los tratamientos. Se muestran dos coordenadas principales a través de DPI 2, 4 y 28, y todos los tipos de muestra (ileal, ápice colónico y contenido fecal). Se utilizó un modelo PERMANOVA para evaluar el efecto del tratamiento sobre la diversidad beta. Los valores p y las estadísticas de R cuadrado se muestran en cada gráfico. Las diferencias significativas se consideraron en base a p < 0,05 (resultados marcados en rojo). El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
Figura 3. Análisis de la diversidad beta de la composición del microbioma GI en grupos de control frente a grupos infectados por Salmonella. Se utilizó la matriz de distancia de Bray-Curtis para calcular la diversidad beta entre los tratamientos. Se muestran dos coordenadas principales a través de DPI 2, 4 y 28, y todos los tipos de muestra (ileal, ápice colónico y contenido fecal). Se utilizó un modelo PERMANOVA para evaluar el efecto del tratamiento sobre la diversidad beta. Los valores p y las estadísticas de R cuadrado se muestran en cada gráfico. Las diferencias significativas se consideraron en base a p < 0,05 (resultados marcados en rojo). El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
Además del modelado basado en PERMANOVA, se utilizó un ANOSIM para evaluar las similitudes de la estructura de la comunidad, y los cambios inducidos por S. Typhimurium fueron significativamente diferentes de otros tratamientos tanto en DPI 4 para el ápice colónico (R = 0.29, p = 0.004) como en DPI 2 para muestras fecales (R = 0.119, p = 0.001), reflejando los resultados de PERMANOVA (p < 0.05) (Figuras suplementarias S5-S13). De acuerdo con los resultados de PERMANOVA, la estrategia de modelado ANOSIM confirmó la ausencia de cambios significativos en la estructura de la comunidad entre los tratamientos para el contenido ileal (p ≥ 0,05). La descomposición adicional de la diversidad beta, analizada por PC1 o PC2 por separado, tampoco mostró cambios significativos en la dispersión de la comunidad para muestras ileales (Figuras suplementarias S14A-F) y confirmó la presencia de alteraciones significativas en la estructura de la comunidad microbiana de los animales infectados por S. typhimurium en DPI 4 para el ápice colónico y DPI 2 para las heces (p < 0.05) (Figuras suplementarias S15, S16A–F). Por último, un análisis transversal de la diversidad beta en muestras fecales demostró diferencias significativas en la estructura de la comunidad para los grupos de S. typhimurium en DPI 7 y 21 (p < 0.05) (Figura suplementaria S17) además de DPI 2 y 4 como se mostró anteriormente. De acuerdo, ANOSIM confirmó los cambios significativos identificados en la estructura de la comunidad en DPI 7 y 21 para muestras fecales en el grupo infectado por S. Typhimurium (p < 0.05) (Figuras suplementarias S18-S21). Por lo tanto, los cambios en los análisis de diversidad alfa y beta ilustran que puede ocurrir un efecto significativo en la estructura de la comunidad durante el cuello de botella inflamatorio en el microbioma gastrointestinal, lo que sugiere una baja capacidad de resistencia, que distinguió de manera única al grupo infectado por S. Typhimurium.
3.4. La red de coocurrencia y el análisis de volatilidad revelaron alteraciones específicas de serovar en la estructura de la comunidad
Además de examinar la estructura de la comunidad a nivel de diversidad beta, se utilizaron redes de co-ocurrencia para evaluar la topología, las interacciones, los taxones centrales y las asociaciones dentro de la comunidad en DPI 2, 4 y 28, tanto para el ápice colónico como para las muestras fecales. Como se observaron cambios mínimos en el intestino delgado, a partir de aquí, la mayor parte del análisis se realizó utilizando el ápice colónico como el sitio primario de inflamación causada por S. Muestras monofásicas y S. Typhimurium, o fecales para examinar las diferencias biogeográficas. En comparación con los grupos infectados, los animales de control mostraron una estructura comunitaria más organizada en DPI 2 y 4 para las muestras de ápice (Figura suplementaria S22 y Figuras 4A-D, respectivamente). Además, la alteración topológica en la arquitectura comunitaria fue más alterada para S. Monofásico y S. typhimurium durante la inflamación (DPI 2 y DPI 4) en comparación con DPI 28 en muestras de ápice, pero no en muestras fecales (Figuras 4A-D y Figuras suplementarias S22-S26). Aunque el tamaño de la muestra para las muestras del ápice colónico fue menor que el de las heces, parece que la arquitectura de la comunidad se distinguió por la biogeografía. Un sello distintivo de la alteración de la red tanto en los taxones centrales como en las asociaciones fue que los productores putativos de ácidos grasos de cadena corta (AGCC) (por ejemplo, Blautia, Faecalibacterium, Alloprevotella, Prevotella, Megasphaera, Dorea, etc.) tenían más probabilidades de estar estrechamente agrupados en los animales de control durante el cuello de botella inflamatorio (DPI 2 y 4) (Figura 4A – grupo de color cian), siendo esa firma más distinguible en el sitio de la inflamación (ápice colónico). Esto sugiere que estos taxones marcan la disbiosis asociada con la infección en todos los serovares probados, pero se acentúa en el caso de S. Monofásico y S. Typhimurium debido a un mayor grado de inflamación. En el caso de S. Monofásica y S. Typhimurium, las alteraciones topológicas de la red de co-ocurrencia se reflejan en una volatilidad potencialmente mayor de la microbiota en el ápice colónico, al examinar el patrón PC1 y la variabilidad en DPI 2, 4 y 28 (Figura suplementaria S27). El análisis de muestras fecales transversales temporales reveló una mayor volatilidad para el microbioma de animales infectados con S. typhimurium (uniformidad variable y cambio en la distribución de PC1 utilizando la matriz de distancia de Bray Curtis) (Figura suplementaria S28). En conjunto, estos resultados apuntan a un efecto de disbiosis en los animales infectados con firmas únicas y compartidas en la estructura de la comunidad.
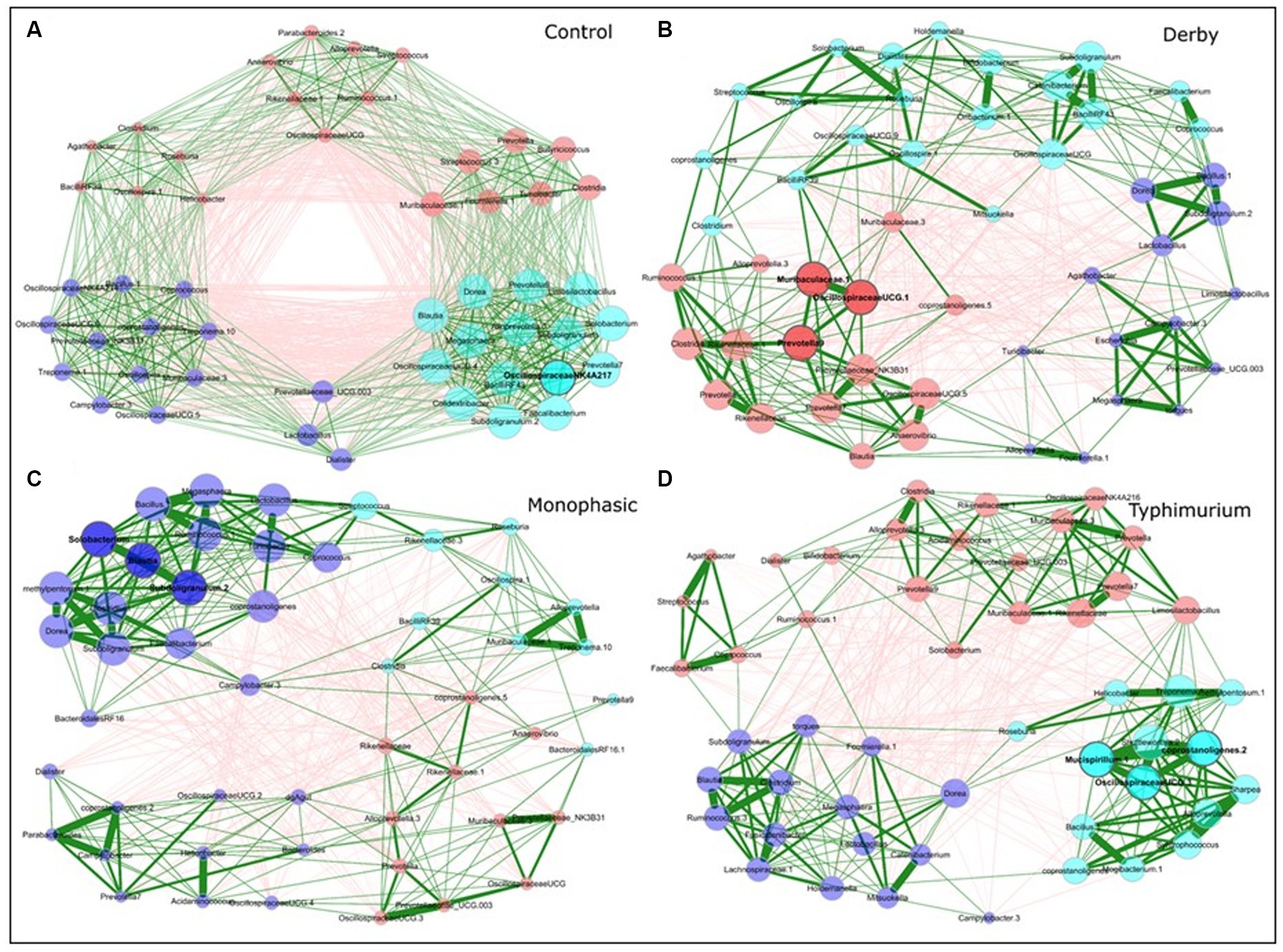 Figura 4. Análisis de red de co-ocurrencia para el microbioma del ápice colónico DPI 4 a través de tratamientos (A – Control; B – Derby; C – monofásica; y D – Typhimurium). Cada nodo representa un taxón bacteriano, y el tamaño de cada nodo se escala de acuerdo con su centralidad. Las asociaciones positivas basadas en Spearman entre taxones se representan como bordes verdes (cuanto más gruesa es la línea, más fuerte es la asociación), mientras que los bordes rojos son indicativos de una anticorrelación entre taxones. No se muestran las aristas que representan un valor inferior a 0,5. Los taxones se colorean en función de los grupos calculados en la construcción de la red. El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
Figura 4. Análisis de red de co-ocurrencia para el microbioma del ápice colónico DPI 4 a través de tratamientos (A – Control; B – Derby; C – monofásica; y D – Typhimurium). Cada nodo representa un taxón bacteriano, y el tamaño de cada nodo se escala de acuerdo con su centralidad. Las asociaciones positivas basadas en Spearman entre taxones se representan como bordes verdes (cuanto más gruesa es la línea, más fuerte es la asociación), mientras que los bordes rojos son indicativos de una anticorrelación entre taxones. No se muestran las aristas que representan un valor inferior a 0,5. Los taxones se colorean en función de los grupos calculados en la construcción de la red. El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
3.5. Los cambios directos en los supuestos productores de SCFA marcan el cuello de botella inflamatorio
A primera vista, el examen taxonómico de animales individuales a través de tratamientos y DPI 2, 4 y 28 demostró un predominio de los filos Firmicutes y Bacteroidetes independientemente de la biogeografía (Figura suplementaria S29). Tras una evaluación más cercana en diferentes niveles jerárquicos de resolución taxonómica (Figuras suplementarias S29-S34), y a pesar de la variación individual esperada, tanto las muestras de ápice colónico como las fecales se enriquecieron ampliamente con los géneros Megasphaera, Prevotella, Alloprevotella, Clostridia y la familia Lachnospiraceae. Un análisis estadístico adicional basado en abundancias relativas reveló taxones de la familia Prevotellaceae como piedra angular y predominantemente afectados por el cuello de botella inflamatorio con variación entre el ápice colónico y las muestras fecales en comparación con el grupo control y entre los serovares (Figuras suplementarias S35-S53). Específicamente, los animales infectados con S. Typhimurium tenían en promedio una proporción significativamente menor de la familia Prevotellaceae (heces en DPI 2 y ápice colónico en DPI 4; Figura suplementaria S35), género Prevotella (heces en DPI 2; Figura suplementaria S36), Prevotellaceae NK3B31 (heces en DPI 2; Figura suplementaria S39), Prevotellaceae UCG 003 (heces en DPI 2 y 4), y Fusicatenibacter (ápice en DPI 2; Figura suplementaria S46); mientras que Megasphaera aumentó en el ápice colónico en DPI 4 y las heces en DPI 2 (p < 0.05) (Figura suplementaria S47). En el caso de S. Monofásica, la proporción de la familia Prevotellaceae se incrementó en el ápice colónico en DPI 4 (Figura Suplementaria S35) y se asoció con una mayor proporción de los géneros Alloprevotella y Prevotellaceae UCG 003 (p < 0,05) (Figuras Suplementarias S37, S41S1, respectivamente). Al igual que S. Typhimurium, el género Fusicatenibacter fue significativamente menor en proporción para S. Monofásico y S. Derby en el ápice colónico en DPI 2 (p < 0,05) (Figura suplementaria S46).
El análisis LEfSe corroboró aún más los resultados del ANOVA para las diferencias en las proporciones, y aunque los resultados deben interpretarse con cautela debido al pequeño tamaño de la muestra, y sugirió una disminución en Clostridia y Lachnospiraceae en el contenido del ápice en DPI 2 y 4 para los animales infectados (Figura 5). La Figura 6 muestra la distribución media de los productores de AGCC putativos más alterados asociados con el cuello de botella inflamatorio al comparar DPI y biogeografía. Las variantes únicas del género Prevotella mostraron una variación específica de serovar distinguible a través del ápice colónico y las heces (por ejemplo, Prevotella 9 y Prevotellacea NK3B31 con mayores abundancias en las heces). Prevotella 9 se encontró en proporción significativamente menor en todos los grupos infectados en DPI 4, pero solo en el ápice colónico (Figura 7). También se analizaron otros taxones clave del microbioma porcino como Lactobacillus, Streptococcus, Campylobacter hyointestinalis y Mitsuokella (Figuras suplementarias S50-S53). Cabe destacar que el género Lactobacillus fue significativamente mayor en S. Typhimurium y Streptococcus fue mayor en proporción en S. Derby infectó animales en múltiples puntos temporales, pero solo en muestras fecales (Cifras suplementarias S50, S51) (p < 0.05). Para el contenido ileal no hubo cambios significativos en las proporciones de taxones entre los tratamientos (p ≥ 0,05), excepto para Alloprevotella en DPI 4 (Figuras suplementarias S54-S73). En conjunto, estos hallazgos sugieren que el cuello de botella inflamatorio más fuerte presente en S. Los animales monofásicos e infectados con S. Typhimurium condujeron a cambios compartidos y únicos en la familia Prevotellaceae o sus representantes, con diferentes firmas biogeográficas subyacentes a la resistencia comunitaria a la infección. Cabe destacar que se puede observar una alta resiliencia en todos esos taxones para muestras de ápice colónico y fecales al examinar el muestreo de punto final, aunque la diferencia en el tamaño de la muestra debe considerarse cuidadosamente en la interpretación.
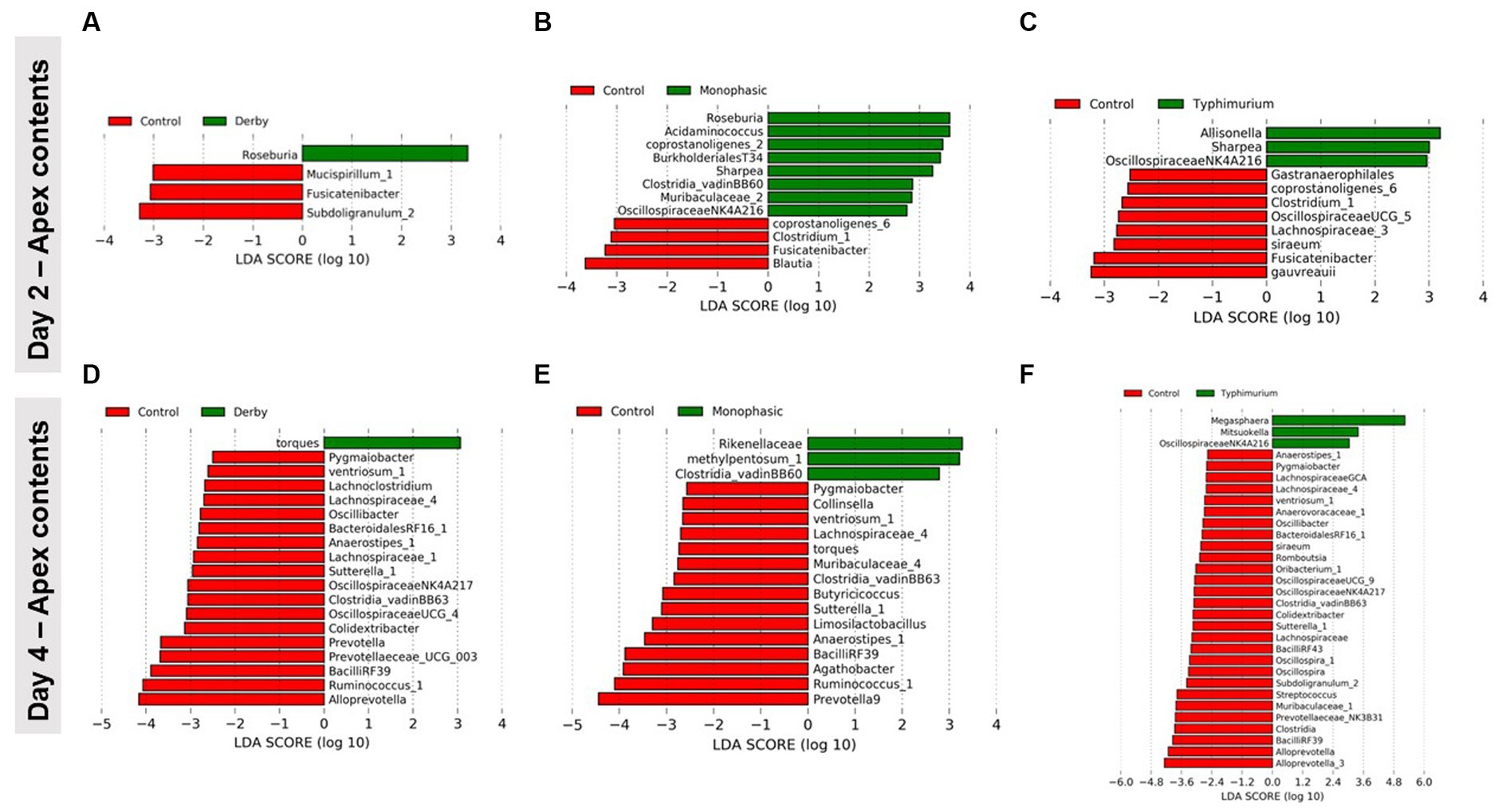 Figura 5. Resultados del análisis LEfSe que demuestran los taxones más diferenciadores entre cada grupo infectado por Salmonella y los animales control. Este análisis se realizó para el contenido del ápice colónico (ápice) solo en DPI 2 (A-C) y 4 (D-F), durante el pico del cuello de botella inflamatorio en la estructura y composición de la comunidad de la microbiota GI. El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
Figura 5. Resultados del análisis LEfSe que demuestran los taxones más diferenciadores entre cada grupo infectado por Salmonella y los animales control. Este análisis se realizó para el contenido del ápice colónico (ápice) solo en DPI 2 (A-C) y 4 (D-F), durante el pico del cuello de botella inflamatorio en la estructura y composición de la comunidad de la microbiota GI. El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
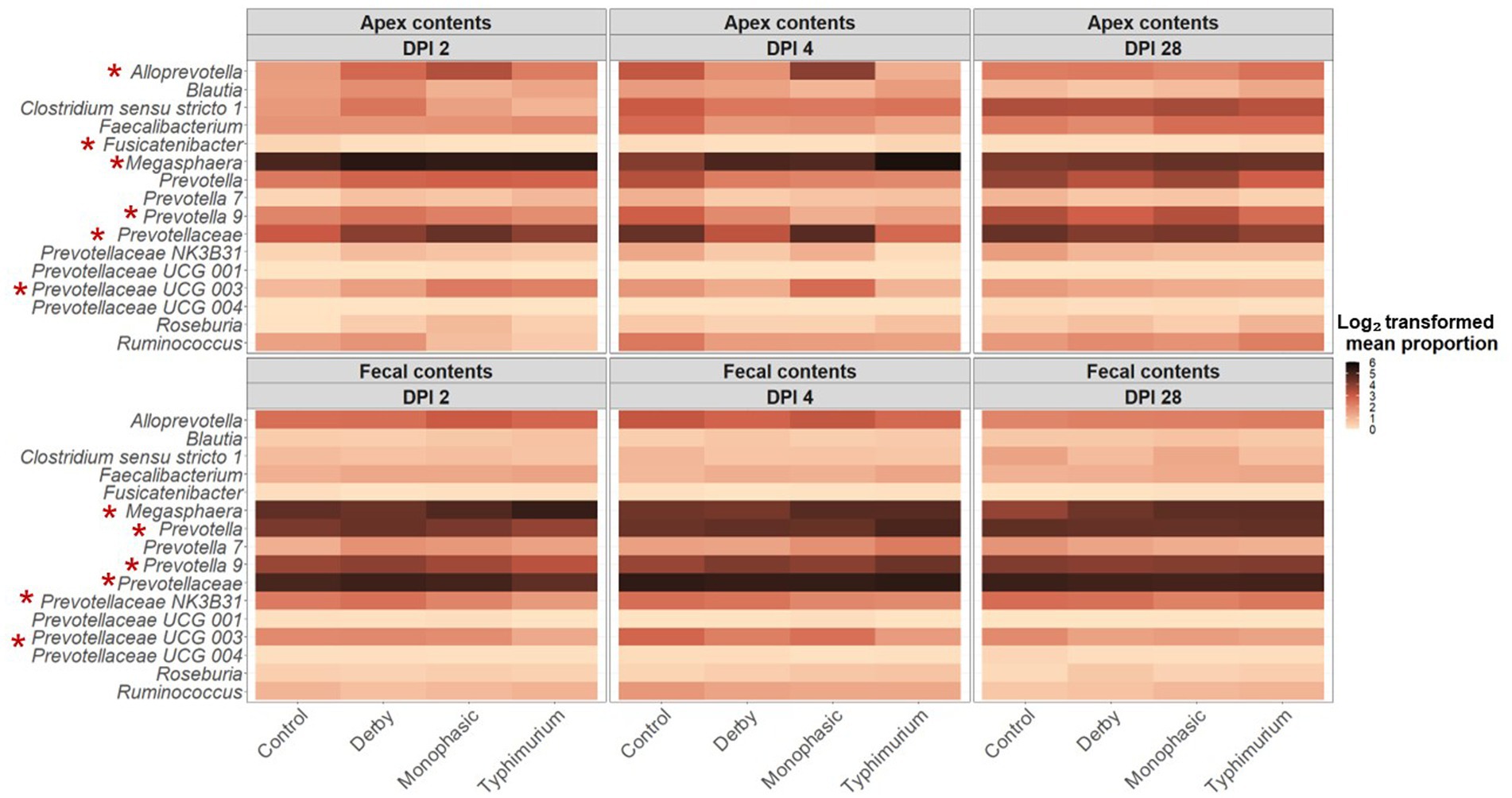 Figura 6. Patrón temporal y biogeográfico de la composición de taxones de microbiomas a través de tratamientos (Control, Derby, Monofásico y Typhimurium). Registro2-proporción media transformada de los productores bacterianos de AGCC putativos más diferenciadores entre los tratamientos. Cuanto más fuerte es el color, más abundante es un taxón dado, en promedio, en todos los tratamientos a lo largo del tiempo. Los asteriscos rojos marcan taxones que fueron significativamente diferentes en proporción entre los tratamientos para DPI 2 o 4, según el tipo de muestra (ápice colónico vs. heces). El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
Figura 6. Patrón temporal y biogeográfico de la composición de taxones de microbiomas a través de tratamientos (Control, Derby, Monofásico y Typhimurium). Registro2-proporción media transformada de los productores bacterianos de AGCC putativos más diferenciadores entre los tratamientos. Cuanto más fuerte es el color, más abundante es un taxón dado, en promedio, en todos los tratamientos a lo largo del tiempo. Los asteriscos rojos marcan taxones que fueron significativamente diferentes en proporción entre los tratamientos para DPI 2 o 4, según el tipo de muestra (ápice colónico vs. heces). El número de animales utilizados por tratamiento puede consultarse en la figura suplementaria S1.
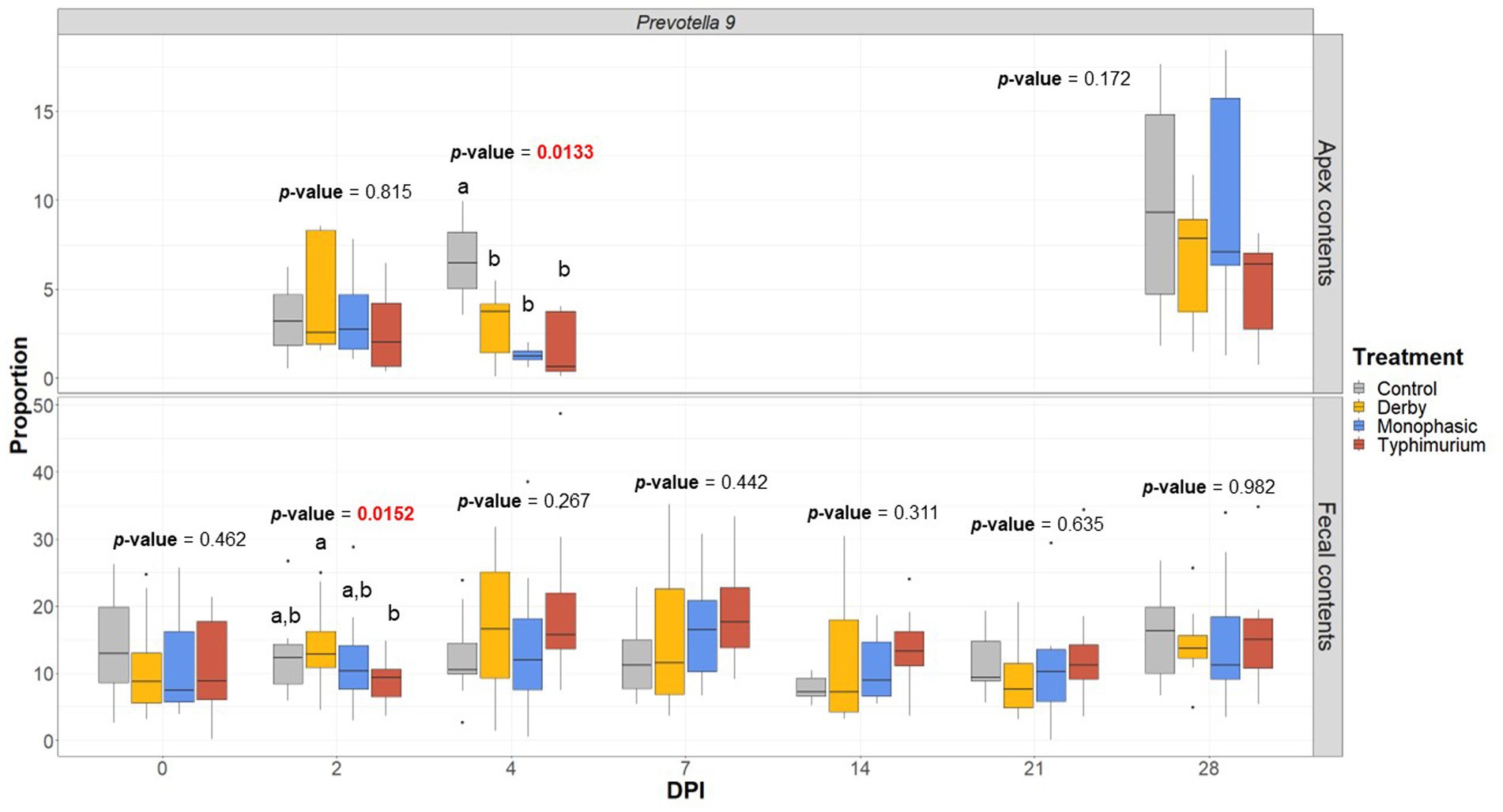 Figura 7. Proporción de Prevotella 9 entre los tratamientos (Control, Derby, Monofásico y Typhimurium) y DPI tanto para el ápice colónico (ápice) como para el contenido fecal. El análisis estadístico se realizó mediante un ANOVA seguido de una prueba T por pares (p < 0,05). Solo se incluyeron en este análisis los animales que tenían muestras de microbioma pasadas a través del límite bioinformático para el control de calidad. Diferentes letras en superíndice indican diferencias significativas entre los tratamientos. La distribución muestral entre los tratamientos y DPI se puede encontrar en la Figura Suplementaria S1.
Figura 7. Proporción de Prevotella 9 entre los tratamientos (Control, Derby, Monofásico y Typhimurium) y DPI tanto para el ápice colónico (ápice) como para el contenido fecal. El análisis estadístico se realizó mediante un ANOVA seguido de una prueba T por pares (p < 0,05). Solo se incluyeron en este análisis los animales que tenían muestras de microbioma pasadas a través del límite bioinformático para el control de calidad. Diferentes letras en superíndice indican diferencias significativas entre los tratamientos. La distribución muestral entre los tratamientos y DPI se puede encontrar en la Figura Suplementaria S1.
3.6. La biogeografía muestral afecta la interpretación de la abundancia de taxones y las asociaciones con las puntuaciones de la enfermedad
Como las bacterias productoras de AGCC putativas eran una marca de disbiosis transitoria inducida por Salmonella y baja resistencia basada en la comunidad, el siguiente paso en el análisis fue correlacionar su proporción con las puntuaciones histopatológicas, que se utilizaron como un proxy para la gravedad de la inflamación específica del nicho. Las alteraciones histopatológicas se evaluaron para el ápice colónico solo o acumulativamente para el intestino grueso como se informó anteriormente (8) y las asociaciones de enfermedades se extrajeron de la proporción de taxones tanto en el ápice colónico como en las heces para identificar firmas únicas y compartidas con bacterias productoras de AGCC putativas. En general, se encontraron valores de correlación de Pearson más fuertes al comparar el ápice colónico frente al microbioma fecal como entrada de datos en el pico de inflamación (DPI 2 y 4) (Figuras 8A-D). Para el microbioma del ápice colónico, solo se encontraron anticorrelaciones significativas con las puntuaciones de la enfermedad cuando se utilizó la puntuación acumulativa del intestino grueso en DPI 4 (Figuras 8A, B), mientras que las correlaciones negativas fueron más fácilmente detectables con el microbioma fecal para ambas S. Monofásico y S. typhimurium (p < 0,05) (Figuras 8C,D). La interpretación de los datos específicamente para el ápice colónico está limitada por el pequeño tamaño de la muestra en DPI 2 y 4. Sin embargo, en DPI 4, para ambos S. Monofásico y S. typhimurium, hubo un patrón general de correlación negativa entre los productores de AGCC putativos no Prevotella cuantificados de manera más consistente en el sitio de la inflamación (Figuras 8A, B) en lugar de con muestras de microbiota fecal (Figuras 8C, D). En el caso de los miembros de la familia Prevotellaceae, específicamente en DPI 4, las correlaciones negativas con las puntuaciones de la enfermedad solo se pudieron encontrar para S. Animales infectados monofásicos cuando se utiliza microbioma fecal (Figuras 8C, D). Para S. Typhimurium, la asociación con los miembros de Prevotellaceae varió a través de la biogeografía y el sistema de puntuación (Figuras 8A-D). En el caso de S. Derby, los taxones de Prevotellaceae a menudo se encontraron anticorrelacionados con el fenotipo de la enfermedad, aunque los valores significativos fueron limitados (p < 0,05). En general, se confirmó que las bacterias productoras de AGCC putativas eran un sello distintivo de la disbiosis inducida por Salmonella (cuello de botella inflamatorio), pero con patrones variables según la biogeografía y el sistema de puntuación de la enfermedad. Finalmente, a pesar de la inoculación directa, la inducción de la enfermedad clínica y el cultivo de Salmonella a partir de heces y contenido intestinal, no se observaron cambios en la prevalencia de Enterobacterales (Figura Suplementaria S74) y la abundancia general fue baja.
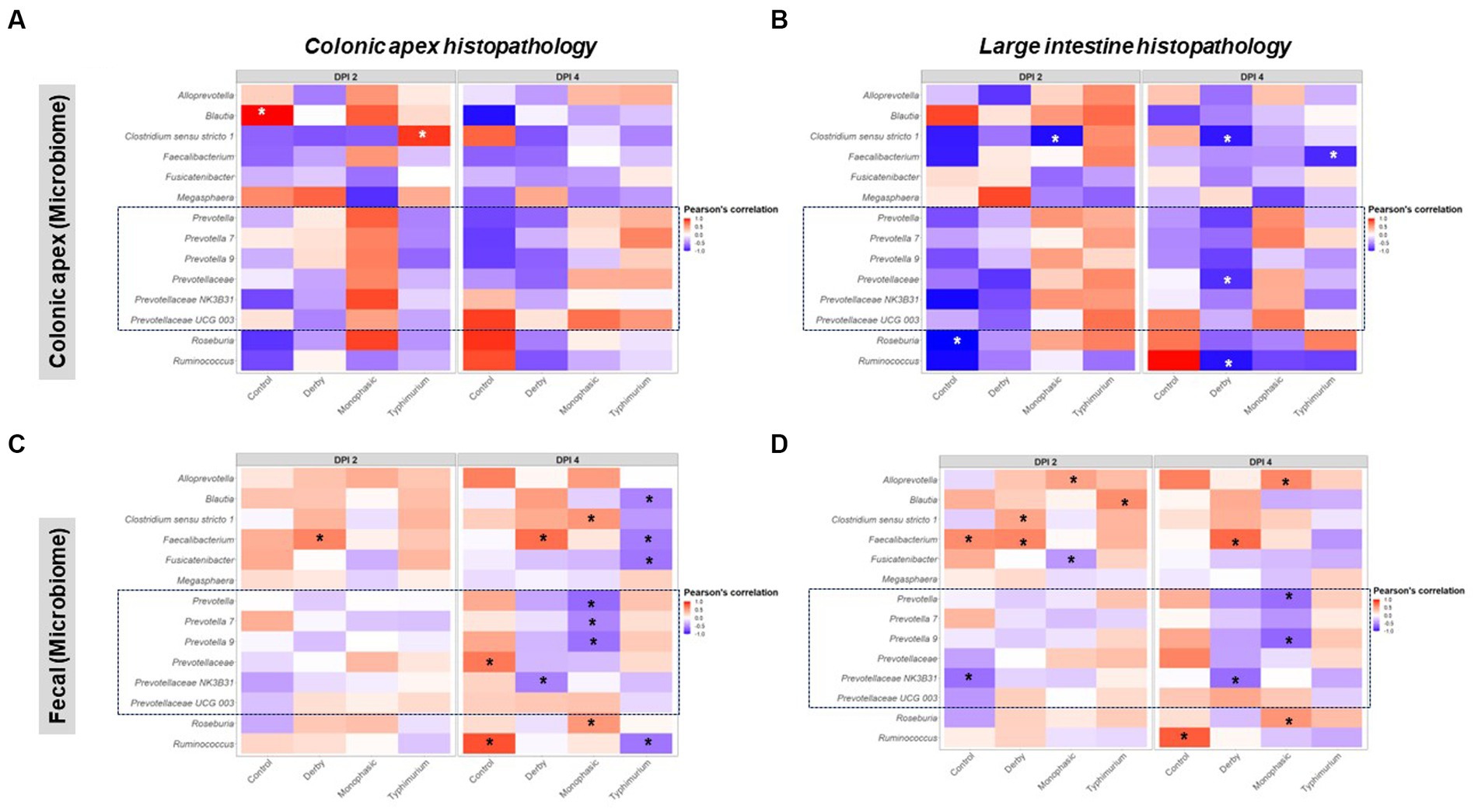 Figura 8. Distribución de correlación de Pearson entre los taxones SCFA putativos y el fenotipo de la enfermedad medido por puntuaciones histopatológicas en el intestino grueso. Las gráficas (A, B) muestran correlaciones entre los taxones SCFA encontrados en las muestras del ápice colónico (ápice) con puntuaciones histopatológicas medidas en el ápice o como una amalgama de secciones del intestino grueso. Del mismo modo, las gráficas (C, D) representan las mismas correlaciones, pero ahora usan distribuciones taxonómicas que se encuentran en el contenido fecal. Todas las correlaciones se calcularon utilizando el logaritmo2 transformación de la proporción total calculada por taxón. El número de animales por tratamiento puede consultarse en la figura suplementaria S1. Los asteriscos sólo están presentes en taxones que pasaron el límite de significación estadística (p < 0,05).
Figura 8. Distribución de correlación de Pearson entre los taxones SCFA putativos y el fenotipo de la enfermedad medido por puntuaciones histopatológicas en el intestino grueso. Las gráficas (A, B) muestran correlaciones entre los taxones SCFA encontrados en las muestras del ápice colónico (ápice) con puntuaciones histopatológicas medidas en el ápice o como una amalgama de secciones del intestino grueso. Del mismo modo, las gráficas (C, D) representan las mismas correlaciones, pero ahora usan distribuciones taxonómicas que se encuentran en el contenido fecal. Todas las correlaciones se calcularon utilizando el logaritmo2 transformación de la proporción total calculada por taxón. El número de animales por tratamiento puede consultarse en la figura suplementaria S1. Los asteriscos sólo están presentes en taxones que pasaron el límite de significación estadística (p < 0,05).
4. Discusión
A pesar de las importantes inversiones en diagnóstico, vigilancia, vacunación, uso de antibióticos y alternativas a los antibióticos (por ejemplo, aditivos para piensos), la salmonelosis sigue siendo una preocupación importante tanto para la medicina porcina como para la humana, especialmente con la aparición de aislados resistentes a múltiples fármacos entre los serovares probados aquí (22, 24, 28, 49-52). En lo que respecta a las estrategias basadas en la dieta, dado que la resistencia a la colonización a múltiples serovares puede no ocurrir a través de un solo mecanismo impulsado por un microorganismo, queda por entender qué especies bacterianas y linajes dentro de una especie funcionarían más eficazmente como probióticos adaptados al huésped para disminuir la prevalencia de Salmonella a nivel de granja (32). El objetivo principal de este estudio fue mapear a nivel de resolución de ARNr 16S la cinética específica de los cambios en los taxones del microbioma GI en el intestino delgado y grueso en las etapas agudas y de recuperación de la inflamación en tres serovares principales de Salmonella (Derby, Monofásico y Typhimurium) que se sabe que residen en el tracto gastrointestinal de los cerdos. Al evaluar los cambios del microbioma gastrointestinal en diferentes sitios anatómicos, serovares y múltiples DPI, pudimos identificar firmas disbióticas específicas relacionadas con la baja resistencia a la perturbación inflamatoria causada por la infección por Salmonella. Específicamente, identificamos firmas compartidas y específicas de serovar previamente demostradas asociadas con la producción de AGCC que se observaron más claramente en el sitio de inflamación (es decir, intestino grueso) y heces.
Al igual que con otros mamíferos, el microbioma porcino tiene características de composición biogeográficamente distintas. En un metaanálisis, Holman et al. (36) demostraron que a nivel de Phylum, el microbioma gástrico e intestinal delgado estaba compuesto principalmente por Firmicutes, mientras que a nivel del intestino grueso, era una composición mixta de predominantemente Bacteroidetes y Firmicutes particularmente enriquecidos con Prevotella (36). Los mismos autores también concluyeron que una fracción de la microbiota se comparte en un mínimo del 90% de las muestras GI, incluyendo Clostridium, Blautia, Lactobacillus, Prevotella, Ruminococcus, Roseburia y otros (36). Trabajos más recientes han corroborado algunos de estos hallazgos al destacar los Clostridiales y Prevotella como parte del microbioma central de lechones de 21 a 35 días (53, 54); esto es consistente con nuestros hallazgos sobre DPI 0 (preinoculación). Sin embargo, el trabajo reciente de Luo et al. (37) demostró una alta variabilidad en la proporción de Prevotellaceae/Prevotella post-destete, creando potencialmente múltiples enterotipos en la población porcina (37). La variabilidad en los enterotipos podría contribuir a la variabilidad de la resistencia comunitaria a la inflamación inducida por Salmonella, como se sugiere en nuestro estudio. Esta ventana de tiempo antes y después del destete es particularmente importante en los sistemas de producción, ya que se predice que será un mayor tiempo de susceptibilidad a la colonización por Salmonella, ya que se ha demostrado que S. typhimurium explota el medio inflamatorio del huésped durante esta ventana para obtener acceso epitelial transintestinal y persistir en cerdos (8, 32, 38, 55, 56). Sobre la base de modelos murinos, se ha demostrado además que S. typhimurium provoca inflamación del huésped, que por consecuencia altera el microambiente de oxígeno (aumento de la aerobiosis) cerca del epitelio, y reduce la población y la diversidad de bacterias sensibles al oxígeno, como los clostridiales, y probablemente otros productores de AGCC que dependen del metabolismo anaeróbico, facilitando así la floración de patógenos (38, 39, 57 ). En condiciones fisiológicas, los AGCC como el butirato se pueden utilizar como fuente de energía para las células epiteliales a través del metabolismo aeróbico. En un microambiente disbiótico (por ejemplo, sitio de inflamación como el ápice colónico), se predice que las concentraciones más bajas de AGCC conducirán a un cambio en el metabolismo de las células epiteliales, que ahora consumen más glucosa como su principal fuente de energía, y por lo tanto aumentan la concentración de oxígeno y óxido nítrico, lo que resulta en la expansión de Enterobacterales (por ejemplo, S. Typhimurium) (39, 58 y 59). La descomposición de este rasgo complejo, a saber, la resistencia a la colonización, sugiere que la presencia tanto de productores de AGCC como de consumidores beneficiosos de oxígeno podría alterar el desprendimiento y la persistencia de S. typhimurium al aumentar la resistencia y la resiliencia basadas en la comunidad a lo largo del tiempo. Se ha demostrado recientemente que existe una anticorrelación entre la abundancia relativa de Prevotella (un taxón clave en el tracto gastrointestinal de los cerdos) y la excreción fecal de ambos S. Monofásico y S. typhimurium en cerdos (33, 34). Prevotella se ha asociado con una mayor producción de AGCC, aunque el AGCC exacto producido por las especies que habitan en el tracto gastrointestinal porcino no ha sido bien caracterizado (34). Aunque carece de la base mecanicista, nuestro trabajo sugiere además que el enriquecimiento del microbioma gastrointestinal con bacterias que pueden alterar la producción de AGCC puede tener un efecto pleiotrópico en la disminución de la patología intestinal.
Como se informó anteriormente, la cantidad media de Salmonella derramada en las heces de los animales en nuestro estudio alcanzó un máximo de aproximadamente 3.0 log10 UFC/ml en DPI 2 a 4 para todos los serovares, después de lo cual la diseminación disminuyó constantemente y no fue detectable en todos menos dos animales infectados con S. typhimurium por DPI 28 (8). A pesar de la inoculación directa, el cultivo de organismos viables de múltiples sitios y la capacidad demostrada para inducir enfermedades clínicas en cerdos para dos de los tres serovares utilizados, la detección de Salmonella en sí no se observó utilizando la metodología 16S empleada aquí en las muestras fecales o en las muestras intestinales recolectadas en la necropsia. La región V4 del ARNr 16S ha demostrado previamente tener una resolución más baja entre géneros (60), por lo tanto, es probable que las lecturas asociadas con Salmonella se asignaran a otros miembros de Enterobacteriacae, sin embargo, tampoco se observaron cambios significativos dentro de este taxón para ningún DPI o tipo de muestra. Esto indica que los cambios sutiles en el microbioma a través de la introducción de organismos patógenos pueden tener impactos significativos en la salud de los animales y el estado de transmisión de enfermedades. Mientras que la enfermedad clínica sólo estaba presente en el S. Typhimurium y S. Grupos monofásicos y no el S. Grupo Derby, nuestro estudio sugiere además que Prevotella puede tener un impacto pleiotrópico en la resistencia a la colonización contra S. Derby, S. Monofásica, y S. Typhimurium, aunque variando los tipos de Prevotellaceae (ASV) tuvieron un efecto biogeográfico distinguible en ambos S. Monofásico y S. typhimurium. Tanto de nuestro estudio como de estudios anteriores, no está claro qué especies específicas de Prevotella podrían tener propiedades distinguibles de resistencia a la colonización; esta limitación se debe nuevamente en parte a los problemas inherentes con el análisis de ARNr 16S, que no proporciona suficiente resolución para una clasificación precisa a nivel de especie (35, 61). Teniendo en cuenta que S. La infección monofásica y por S. typhimurium y la inflamación pueden compartimentarse en segmentos específicos del intestino grueso (por ejemplo, tejidos colónicos ciegos y ápice) (8), se necesitan más estudios para evaluar la composición de las comunidades microbianas en esos sitios anatómicos específicos, incluida una comparación de las comunidades luminales a las asociadas a la mucosa (que se intentó aquí pero fracasó probablemente debido al bajo rendimiento y la contaminación con ADN del huésped), el uso de técnicas de secuenciación más profundas (es decir, secuenciación de escopeta) y el aislamiento y la secuenciación del genoma completo de especies autóctonas de Prevotella con propiedades probióticas demostrables in vivo (35). Como nuestro estudio tenía un poder estadístico limitado y no midió las concentraciones de AGCC, se podrían hacer esfuerzos posteriores para tener una cuantificación absoluta más sólida de microorganismos y metabolitos potencialmente protectores tanto en el sitio de la infección como en las heces.
Siempre que se encuentre que especies específicas de Prevotella sp. tienen propiedades anti-transporte consistentes contra S. Monofásica y S. Typhimurium, la genética / genómica bacteriana combinada con la experimentación animal in vivo podría permitir el sondeo de rasgos metabólicos únicos que podrían aprovecharse para enriquecer a tales especies utilizando cambios en la dieta en las granjas (32, 35). Entre las especies conocidas, Prevotella copri y P. stercorea son las especies candidatas más probables para tener un efecto protector sobre la infección por Salmonella en cerdos (35). Como prueba de concepto, Trachsel et al. (33) demostraron recientemente que la incorporación de almidón de patata resistente en la dieta de los cerdos para enriquecer especies de interés (por ejemplo, Prevotella sp.) en el sitio de la infección podría ayudar a reducir el transporte de S multirresistente. Monofásico; La alimentación directa de ácidos grasos asociados con la resistencia a las enfermedades no produjo un efecto positivo (33). Estudios adicionales han corroborado la hipótesis de que la utilización de almidón resistente puede alterar directamente la composición del microbioma intestinal porcino a través del enriquecimiento de bacterias SCFA como los filotipos asociados a Prevotella, Lachnospiraceae y Ruminococcus (62, 63). La utilización de fibras altamente fermentables puede incluso tener un efecto más amplio en la salud intestinal, ya que es beneficioso contra otro patógeno porcino IG, Brachyspira hyodysenteriae (64). Alternativamente, el salvado de maíz puede ser una fuente de fibra capaz de modular el microbioma GI porcino al inducir microbios SCFA que se plantearía la hipótesis de que tienen efectos positivos en términos de reducir el desprendimiento de S. enterica en cerdos (65). Cabe destacar la posible expansión de Alloprevotella en S. Los cerdos infectados con S. Typhimurium observados aquí pueden estar asociados con la utilización de lactato por estos microbios (66, 67), que es un metabolito derivado del huésped en la infección por S. Typhimurium en un modelo murino (68, 69). La alimentación directa de ácidos grasos a los cerdos como medida preventiva puede ser un desafío debido a los efectos variables de diferentes moléculas, la identificación de concentraciones biológicas efectivas sin alterar la palatabilidad de la dieta, la absorción y excreción GI variable, los posibles efectos impredecibles en otros miembros beneficiosos de la comunidad microbiana y la influencia del sistema inmune del huésped (70). Por lo tanto, las estrategias de alimentación con ingredientes como el almidón de patata resistente que enriquecen directamente para especies de interés (por ejemplo, Prevotella sp.) en el sitio de inflamación (33), o la incorporación directa de probióticos comercialmente viables con las propiedades deseadas, tienen más probabilidades de ser eficaces para mejorar la resistencia a la colonización y disminuir el transporte de Salmonella en granjas porcinas. Dependiendo de la mezcla de probióticos (por ejemplo, Bacillus amyloliquefaciens G10, Levilactobacillus brevis M10 y Limosilactobacillus reuteri RTR), puede haber una reducción en la abundancia relativa de productores de AGCC en el tracto gastrointestinal de los cerdos (66), lo que se plantearía la hipótesis de que disminuiría la resistencia a la colonización contra los serovares probados aquí. Además, no se sabe cómo la variación de la cepa de Prevotella puede influir en el ensamblaje del microbioma del intestino grueso del cerdo y, por consiguiente, afectar la varianza en la resistencia a la colonización contra Salmonella. Se ha informado que dicha diversidad genética existe en P. copri aislado de humanos y se predice que se ha visto afectada por la dieta, ya que la disminución del contenido de fibra es un factor perjudicial potencial para el injerto del microbioma gastrointestinal (71). Además, se ha informado recientemente que los distintos aislados de P. copri pueden utilizar mejor ciertos polisacáridos dietéticos que pueden convertirse en una base para el estudio de nuevos enfoques simbióticos (72). Una nota de advertencia es que no todas las Prevotella pueden tener efectos beneficiosos, pero bajo susceptibilidad genética específica del huésped podrían comportarse como un patobiológico (73). Otros posibles factores de confusión, como el micobioma GI porcino, también pueden influir en la capacidad de Prevotella para disminuir el transporte de Salmonella, ya que estudios recientes de Summers et al. apuntan a la existencia de un hongo beneficioso autóctono en el intestino grueso porcino, Kazachstania slooffiae, que se correlaciona positivamente con la abundancia de Prevotella sp. (53, 54, 74 y 75). Por lo tanto, otro componente de este rasgo complejo, a saber, la resistencia a la colonización contra Salmonella en cerdos, podría estar parcialmente mediada por una interacción mutualista entre bacterias y levaduras en el tracto gastrointestinal.
A pesar de incluir múltiples serovares en este estudio en un intento de evaluar el efecto de diferentes serovares en la composición del microbioma, se sabe que la población de S. Derby, S. Monofásico y S. typhimurium son genéticamente diversos tanto en el tipo de secuencia (ST) como en los niveles de cgMLST (4, 11, 12, 16, 50, 76). Cada serovar tiene un alto grado de clonalidad a nivel ST (muy pocas ST dominantes distribuidas en todo el mundo), y se eligieron aislados de los grupos ST dominantes para permitir las comparaciones (Typhimurium, ST19; Monofásica, ST34, Derby, ST40) (8); En el futuro se deben emplear enfoques experimentales adicionales para tener en cuenta la diversidad filogenética y la variación en el potencial zoonótico (4, 12). El uso de una combinación de ST de un serovar dado para evaluar las propiedades de resistencia a la colonización del microbioma GI o un cóctel específico de probióticos candidatos determinaría de manera más sólida su eficacia. Cuantos más ST y cgMLST se incluyan en cada cóctel específico de serovar que se probará, más probable es que la estrategia probiótica o de intervención sea generalizable a nivel de granja. Además, la variación en los patrones de resistencia a los antimicrobianos (RAM) específicos de serovar dentro de las estructuras de población (ST y cgMLST) podría incluirse en la evaluación de las intervenciones basadas en el microbioma que podrían aplicarse potencialmente a los linajes de RAM.
En resumen, este estudio demuestra la importancia de estudiar los cambios biogeográficos del microbioma GI para aprovechar la información sobre las propiedades específicas del microbioma que pueden ayudar en la resistencia a la colonización contra los serovares zoonóticos del linaje I de S. enterica que residen en cerdos. También subraya el hecho de que el monitoreo fecal de la diversidad del microbioma gastrointestinal o los taxones no siempre puede ser predictivo de la resistencia a la colonización. Por último, nuestro trabajo destaca una firma disbiótica de cerdos infectados con Salmonella que altera principalmente la composición y la proporción de productores putativos de AGCC que se relacionó con el cuello de botella inflamatorio creado por el patógeno. De ellos, Prevotella fue identificado como un taxón clave en el microbioma GI porcino que parece ser un biomarcador con potencial para tener un impacto en el fenotipo de la enfermedad, tal vez como un taxón central para mejorar la resistencia comunitaria a la infección. Por lo tanto, nuestro trabajo, junto con otros, apunta hacia un efecto específico de nicho relacionado con los cambios en el microbioma en el sitio de la infección; esta información se puede utilizar para informar el desarrollo futuro de estrategias probióticas, prebióticas o simbióticas para el control escalable de Salmonella en granjas a nivel estrecho (específico de serovar) o de amplio espectro.
Declaración de disponibilidad de datos
Los conjuntos de datos presentados en este estudio se pueden encontrar en repositorios en línea. Los nombres del repositorio o repositorios y los números de acceso se pueden encontrar en: https://www.ncbi.nlm.nih.gov/bioproject/944612.
Declaración ética
Los estudios en animales fueron aprobados por el Comité Institucional de Cuidado y Uso de Animales de la Universidad Estatal de Iowa. Los estudios se realizaron de acuerdo con la legislación local y los requisitos institucionales. No se obtuvo el consentimiento informado por escrito de los propietarios para la participación de sus animales en este estudio porque los animales eran animales de investigación propiedad de la universidad, por lo tanto, el consentimiento informado no era aplicable.
Contribuciones del autor
AK y BA concibieron y diseñaron el proyecto. SN fue responsable de realizar los estudios experimentales con la ayuda de AK, BA y otros (ver «Agradecimientos»). SN preparó muestras para análisis metagenómico y BA realizó las evaluaciones histopatológicas. JCG-N, NP y AK realizaron todos los análisis de datos metagenómicos y generaron todas las visualizaciones de datos. NP llevó a cabo todo el trabajo de bioinformática. JCG-N, NP y AK escribieron el manuscrito inicial. NK y AB ayudaron con la interpretación de los datos y la preparación final del manuscrito. NK lideró el análisis de la red y LEfSe, incluida la preparación de figuras y la interpretación de datos. Todos los autores contribuyeron al artículo y aprobaron la versión presentada.
Financiación
Este trabajo fue apoyado en parte por fondos proporcionados por la National Pork Board (subvención # 16-215: Investigación de patogenicidad, aptitud competitiva y métodos novedosos para el diagnóstico rápido de S. 4,[5],12:i:-).
Reconocimientos
Los autores desean agradecer a Paulo Arruda, Eric R. Burrough, Drew R. Magstadt, Franco Matias Ferreyra, Igor RH Gatto y Henrique Meiroz de Souza Almeida por su ayuda con la recolección de muestras durante los estudios en animales y Paul J. Plummer por su ayuda con el análisis preliminar de datos metagenómicos. También nos gustaría agradecer a Edward Deehan por sus comentarios constructivos sobre las revisiones de manuscritos y el ajuste en la presentación de datos. Este trabajo se completó utilizando el Holland Computing Center de la Universidad de Nebraska, que recibe apoyo de la Iniciativa de Investigación de Nebraska.
Conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un posible conflicto de intereses.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o reclamo que pueda ser hecho por su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fvets.2023.1186554/full#supplementary-material
Notas
1. ^https://hcc.unl.edu/
Referencias
1. CDC. (2021). Seguridad alimentaria. Disponible en: https://www.cdc.gov/foodsafety/foodborne-germs.html.
3. Ferrari, RG, Rosario, DKA, Cunha-Neto, A, Mano, SB, Figueiredo, EES, y Conte-Junior, CA. Worldwide epidemiology of Salmonella serovars in animal-based foods: a meta-analysis. Aplicación Environ Microbiol. (2019) 85:E00591–19. doi: 10.1128/AEM.00591-19
Resumen de PubMed | Texto completo de CrossRef | Google Académico
4. Achtman, M, Wain, J, Weill, F-X, Nair, S, Zhou, Z, Sangal, V, et al. Tipificación de secuencias multilocus como reemplazo del serotipado en Salmonella enterica. PLoS Pathog. (2012) 8:E1002776. doi: 10.1371/journal.ppat.1002776
Resumen de PubMed | Texto completo de CrossRef | Google Académico
5. Hauser, E, Hebner, F, Tietze, E, Helmuth, R, Junker, E, Prager, R, et al. Diversidad de Salmonella enterica serovar Derby aislada de cerdo, cerdo y humanos en Alemania. Int J Food Microbiol. (2011) 151:141–9. doi: 10.1016/j.ijfoodmicro.2011.08.020
Resumen de PubMed | Texto completo de CrossRef | Google Académico
6. Kerouanton, A, Rose, V, Weill, F-X, Granier, SA y Denis, M. Diversidad genética y perfiles de resistencia antimicrobiana de Salmonella enterica serotipo Derby aislado de cerdos, cerdos y humanos en Francia. Foodborne Pathog Dis. (2013) 10:977–84. DOI: 10.1089/FPD.2013.1537
Resumen de PubMed | Texto completo de CrossRef | Google Académico
7. Sanchez, J, Dohoo, IR, Christensen, J, and Rajic, A. Factors influencing the prevalence of Salmonella spp. in swine farms: a meta-analysis approach. Prev Vet Med. (2007) 81:148–77. doi: 10.1016/j.prevetmed.2007.04.005
Resumen de PubMed | Texto completo de CrossRef | Google Académico
8. Naberhaus, SA, Krull, AC, Arruda, BL, Arruda, P, Sahin, O, Schwartz, KJ, et al. Patogenicidad y aptitud competitiva de Salmonella enterica serovar 4,[5],12:i:- en comparación con Salmonella Typhimurium y Salmonella Derby en cerdos. Front Vet Sci. (2020) 6:502. DOI: 10.3389/fvets.2019.00502
Resumen de PubMed | Texto completo de CrossRef | Google Académico
9. Peckham, CF, y Savage, WG. Un brote de intoxicación por pastel de cerdo en Derby. J Hyg. (1923) 22:69–76. doi: 10.1017/S0022172400008068
Resumen de PubMed | Texto completo de CrossRef | Google Académico
10. Cui, S, Li, J, Sun, Z, Hu, C, Jin, S, Li, F, et al. Caracterización de aislados de Salmonella enterica de bebés y niños pequeños en Wuhan, China. J Quema antimicrobiana. (2008) 63:87–94. doi: 10.1093/jac/dkn452
11. Gomes-Neto, JC, Pavlovikj, N, Cano, C, Abdalhamid, B, Al-Ghalith, GA, Loy, JD, et al. Minería de población heurística y jerárquica de pangenomas del linaje I de Salmonella enterica como plataforma para mejorar la seguridad alimentaria. Sistema de alimentos de sostenimiento frontal. (2021) 5:725791. DOI: 10.3389/FSUFS.2021.725791
12. Alikhan, N-F, Zhou, Z, Sergeant, MJ, y Achtman, M. Una visión genómica de la estructura poblacional de Salmonella. PLoS Genet. (2018) 14:E1007261. doi: 10.1371/journal.pgen.1007261
Resumen de PubMed | Texto completo de CrossRef | Google Académico
13. Sun, H, Wan, Y, Du, P, y Bai, L. La epidemiología de la Salmonella Typhimurium monofásica. Foodborne Pathog Dis. (2020) 17:87–97. DOI: 10.1089/FPD.2019.2676
14. Moreno Switt, AI, Soyer, Y, Warnick, LD, and Wiedmann, M. Emergence, distribution, and molecular and phenotypic characteristics of Salmonella enterica serotype 4,5,12:i:-. Foodborne Pathog Dis. (2009) 6:407–15. DOI: 10.1089/FPD.2008.0213
Resumen de PubMed | Texto completo de CrossRef | Google Académico
15. Arai, N, Sekizuka, T, Tamamura, Y, Kusumoto, M, Hinenoya, A, Yamasaki, S, et al. La isla genómica 3 de Salmonella es un elemento integrador y conjugativo y contribuye a la tolerancia al cobre y al arsénico de Salmonella enterica. Agentes antimicrobianos Chemother. (2019) 63:E00429–19. doi: 10.1128/AAC.00429-19
Resumen de PubMed | Texto completo de CrossRef | Google Académico
16. Branchu, P, Charity, OJ, Bawn, M, Thilliez, G, Dallman, TJ, Petrovska, L, et al. SGI-4 in monophasic Salmonella Typhimurium ST34 es un nuevo ICE que mejora la resistencia al cobre. Microbiol frontal. (2019) 10:1118. doi: 10.3389/fmicb.2019.01118
Resumen de PubMed | Texto completo de CrossRef | Google Académico
17. Clark, CG, Landgraff, C, Robertson, J, Pollari, F, Parker, S, Nadon, C, et al. Distribución de elementos resistentes a metales pesados en poblaciones canadienses de Salmonella 4,[5],12:i:- y asociación con los genotipos monofásicos y fenotipo. PLoS One. (2020) 15:E0236436. doi: 10.1371/journal.pone.0236436
Resumen de PubMed | Texto completo de CrossRef | Google Académico
18. Mourão, J, Novais, C, Machado, J, Peixe, L, and Antunes, P. Metal tolerance in emerging clinically relevant multidrug-resistant Salmonella enterica serotype 4,[5],12:i:− clones circulating in Europe. Int J Agentes antimicrobianos. (2015) 45:610–6. doi: 10.1016/j.ijantimicag.2015.01.013
19. Bearson, BL, Trachsel, JM, Shippy, DC, Sivasankaran, SK, Kerr, BJ, Loving, CL, et al. El papel de la isla genómica 4 de Salmonella en la tolerancia a metales de Salmonella enterica serovar I 4,[5],12:i:- aislado de brote de carne de cerdo USDA15WA-1. Genes. (2020) 11:1291. doi: 10.3390/genes11111291
Resumen de PubMed | Texto completo de CrossRef | Google Académico
20. He, J, Sun, F, Sun, D, Wang, Z, Jin, S, Pan, Z, et al. Resistencia a múltiples fármacos y prevalencia de genes de resistencia a quinolonas de Salmonella enterica serotipos 4,[5],12:i:- en China. Int J Food Microbiol. (2020) 330:108692. doi: 10.1016/j.ijfoodmicro.2020.108692
Resumen de PubMed | Texto completo de CrossRef | Google Académico
21. Ingle, DJ, Ambrose, RL, Baines, SL, Duchene, S, Gonçalves da Silva, A, Lee, DYJ, et al. Dinámica evolutiva de Salmonella enterica serovar 4,[5],12:i:- resistente a múltiples fármacos en Australia. Nat Commun. (2021) 12:4786. DOI: 10.1038/S41467-021-25073-W
Resumen de PubMed | Texto completo de CrossRef | Google Académico
22. Long, L, You, L, Wang, D, Wang, M, Wang, J, Bai, G, et al. MDR altamente prevalente, frecuentemente portadora de genes de virulencia y genes de resistencia antimicrobiana en aislados de Salmonella enterica serovar 4,[5],12:i:- de la provincia de Guizhou, China. PLoS One. (2022) 17:E0266443. doi: 10.1371/journal.pone.0266443
Resumen de PubMed | Texto completo de CrossRef | Google Académico
23. Pires, J, Huisman, JS, Bonhoeffer, S, y Van Boeckel, TP. Multidrug resistance dynamics in Salmonella in food animals in the United States: an analysis of genomes from public databases. Microbiol Spectr. (2021) 9:E00495–21. doi: 10.1128/Spectrum.00495-21
Resumen de PubMed | Texto completo de CrossRef | Google Académico
24. Qin, X, Yang, M, Cai, H, Liu, Y, Gorris, L, Aslam, MZ, et al. Resistencia a antibióticos de la variante monofásica 1,4,[5],12:i:-en China: una revisión sistemática y metanálisis. Antibióticos. (2022) 11:532. doi: 10.3390/antibióticos11040532
Resumen de PubMed | Texto completo de CrossRef | Google Académico
25. Berge, AC, y Wierup, M. Estrategias nutricionales para combatir la Salmonella en la producción animal de alimentos monogástricos. Animal. (2012) 6:557–64. doi: 10.1017/S1751731111002217
Resumen de PubMed | Texto completo de CrossRef | Google Académico
26. de la Cruz, ML, Conrado, I, Nault, A, Perez, A, Dominguez, L, and Alvarez, J. Vaccination as a control strategy against Salmonella infection in pigs: a systematic review and meta-analysis of the literature. Res Vet Sci. (2017) 114:86–94. doi: 10.1016/j.rvsc.2017.03.005
Resumen de PubMed | Texto completo de CrossRef | Google Académico
27. Young, I, Wilhelm, BJ, Cahill, S, Nakagawa, R, Desmarchelier, P, y Rajić, A. Una revisión sistemática rápida y un metanálisis de la eficacia de las intervenciones de sacrificio y procesamiento para controlar la Salmonella no tifoidea en la carne de res y cerdo. J Food Prot. (2016) 79:2196–210. doi: 10.4315/0362-028X. JFP-16-203
Resumen de PubMed | Texto completo de CrossRef | Google Académico
28. Bearson, SMD. Salmonella en cerdos: prevalencia, resistencia a múltiples fármacos y estrategias de vacunación. Annu Rev Anim Biosci. (2022) 10:373–93. doi: 10.1146/annurev-animal-013120-043304
Resumen de PubMed | Texto completo de CrossRef | Google Académico
29. Davies, PR. Producción porcina intensiva y seguridad porcina. Foodborne Pathog Dis. (2011) 8:189–201. DOI: 10.1089/FPD.2010.0717
Resumen de PubMed | Texto completo de CrossRef | Google Académico
30. Ojha, S, y Kostrzynska, M. Enfoques para reducir la Salmonella en la producción de carne de cerdo. J Food Prot. (2007) 70:2676–94. doi: 10.4315/0362-028X-70.11.2676
31. Helke, KL, McCrackin, MA, Galloway, AM, Poole, AZ, Salgado, CD y Marriott, BP. Efectos del uso de antimicrobianos en animales agrícolas sobre la salmonelosis transmitida por los alimentos resistentes a los medicamentos en humanos: una revisión sistemática de la literatura. Crit Rev Food Sci Nutr. (2017) 57:472–88. doi: 10.1080/10408398.2016.1230088
Resumen de PubMed | Texto completo de CrossRef | Google Académico
32. Kim, HB, e Isaacson, RE. Salmonella en cerdos: interacciones de la microbiota. Annu Rev Anim Biosci. (2017) 5:43–63. doi: 10.1146/annurev-animal-022516-022834
33. Trachsel, JM, Bearson, BL, Kerr, BJ, Shippy, DC, Byrne, KA, Loving, CL, et al. Ácidos grasos de cadena corta y taxones bacterianos asociados con la reducción de Salmonella enterica serovar I 4,[5],12:i:- derramamiento en cerdos alimentados con una dieta suplementada con almidón de patata resistente. Microbiol Spectr. (2022) 10:E02202–21. doi: 10.1128/spectrum.02202-21
Resumen de PubMed | Texto completo de CrossRef | Google Académico
34. Bearson, SMD, Allen, HK, Bearson, BL, Looft, T, Brunelle, BW, Kich, JD, et al. Perfilando la microbiota gastrointestinal en respuesta a Salmonella: baja versus alta Salmonella derramamiento en el huésped porcino natural. Infectar Genet Evol. (2013) 16:330–40. doi: 10.1016/j.meegid.2013.03.022
35. Amat, S, Lantz, H, Munyaka, PM, y Willing, BP. Prevotella en cerdos: las asociaciones positivas y negativas con la producción y la salud. Microorganismos. (2020) 8:1584. doi: 10.3390/microorganismos8101584
Resumen de PubMed | Texto completo de CrossRef | Google Académico
36. Holman, DB, Brunelle, BW, Trachsel, J y Allen, HK. Metaanálisis para definir una microbiota central en el intestino porcino. mSystems. (2017) 2:E00004–17. doi: 10.1128/mSystems.00004-17
37. Luo, Y, Ren, W, Smidt, H, Wright, A-DG, Yu, B, Schyns, G, et al. Distribución dinámica de la microbiota intestinal en cerdos en diferentes fases de crecimiento: composición y contribución. Microbiol Spectr. (2022) 10:E00688:E0068821. doi: 10.1128/spectrum.00688-21
Resumen de PubMed | Texto completo de CrossRef | Google Académico
38. Bescucci, DM, Moote, PE, Ortega Polo, R, Uwiera, RRE, e Inglis, GD. Salmonella enterica serovar Typhimurium modula temporalmente la microbiota entérica y las respuestas del huésped para superar la resistencia a la colonización en cerdos. Aplicación Environ Microbiol. (2020) 86:E01569–20. doi: 10.1128/AEM.01569-20
Resumen de PubMed | Texto completo de CrossRef | Google Académico
39. Olsan, EE, Byndloss, MX, Faber, F, Rivera-Chávez, F, Tsolis, RM y Bäumler, AJ. Resistencia a la colonización: la deconvolución de un rasgo complejo. J Biol Chem. (2017) 292:8577–81. DOI: 10.1074/JBC. R116.752295
Resumen de PubMed | Texto completo de CrossRef | Google Académico
40. Apprill, A, McNally, S, Parsons, R, and Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer increases great detection of SAR11 bacterioplankton. Aquat Microb Ecol. (2015) 75:129–37. DOI: 10.3354/ame01753
41. Caporaso, JG, Lauber, CL, Walters, WA, Berg-Lyons, D, Lozupone, CA, Turnbaugh, PJ, et al. Patrones globales de diversidad de ARNr 16S a una profundidad de millones de secuencias por muestra. Proc Natl Acad Sci U S A. (2011) 108:4516–22. DOI: 10.1073/PNAS.1000080107
Resumen de PubMed | Texto completo de CrossRef | Google Académico
42. Parada, AE, Needham, DM, y Fuhrman, JA. Cada base importa: evaluación de pequeños cebadores de ARNr de subunidades para microbiomas marinos con comunidades simuladas, series temporales y muestras de campo globales: cebadores para estudios de microbiomas marinos. Environ Microbiol. (2016) 18:1403–14. doi: 10.1111/1462-2920.13023
Resumen de PubMed | Texto completo de CrossRef | Google Académico
43. Matsuki, T, Watanabe, K, Fujimoto, J, Takada, T, and Tanaka, R. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Aplicación Environ Microbiol. (2004) 70:7220–8. doi: 10.1128/AEM.70.12.7220-7228.2004
Resumen de PubMed | Texto completo de CrossRef | Google Académico
44. Bolyen, E, Rideout, JR, Dillon, MR, Bokulich, NA, Abnet, CC, Al-Ghalith, GA, et al. Ciencia de datos de microbiomas reproducible, interactiva, escalable y extensible utilizando QIIME 2. Nat Biotechnol. (2019) 37:852–7. DOI: 10.1038/S41587-019-0209-9
Resumen de PubMed | Texto completo de CrossRef | Google Académico
45. Callahan, BJ, McMurdie, PJ, Rosen, MJ, Han, AW, Johnson, AJA, and Holmes, SP. DADA2: high-resolution sample inference from Illumina amplicon data. Métodos Nat. (2016) 13:581–3. DOI: 10.1038/NMETH.3869
Resumen de PubMed | Texto completo de CrossRef | Google Académico
46. Segata, N, Izard, J, Waldron, L, Gevers, D, Miropolsky, L, Garrett, WS, et al. Descubrimiento y explicación de biomarcadores metagenómicos. Genoma Biol. (2011) 12:R60. DOI: 10.1186/GB-2011-12-6-R60
Resumen de PubMed | Texto completo de CrossRef | Google Académico
47. The Galaxy CommunityAfgan, E, Nekrutenko, A, Grüning, BA, Blankenberg, D, Goecks, J, et al. La plataforma Galaxy para análisis biomédicos accesibles, reproducibles y colaborativos: actualización 2022. Ácidos nucleicos res. (2022) 50:W345–51. doi: 10.1093/nar/gkac247
Resumen de PubMed | Texto completo de CrossRef | Google Académico
48. Peschel, S, Müller, CL, von Mutius, E, Boulesteix, A-L, y Depner, M. NetCoMi: network construction and comparison for microbiome data in R. Brief Bioinform. (2021) 22:bbaa290. doi: 10.1093/bib/bbaa290
Resumen de PubMed | Texto completo de CrossRef | Google Académico
49. Sun, R-Y, Ke, B-X, Fang, L-X, Guo, W-Y, Li, X-P, Yu, Y, et al. Diseminación clonal global de MDR ST3 portadora de mcr-34 Salmonella enterica serotipo Typhimurium y variantes monofásicas 1,4,[5],12:i:− de aislados clínicos. J Quema antimicrobiana. (2020) 75:1756–65. doi: 10.1093/jac/dkaa115
50. Bawn, M, Alikhan, N-F, Thilliez, G, Kirkwood, M, Wheeler, NE, Petrovska, L, et al. Evolución del serotipo Typhimurium de Salmonella enterica impulsada por la selección antropogénica y la adaptación al nicho. PLoS Genet. (2020) 16:E1008850. doi: 10.1371/journal.pgen.1008850
Resumen de PubMed | Texto completo de CrossRef | Google Académico
51. Gomes-Neves, E, Antunes, P, Manageiro, V, Gärtner, F, Caniça, M, da Costa, JMC, et al. Salmonella enterica multirresistente clínicamente relevante en manipuladores de carne y cerdos en el matadero. Veterinario Microbiol. (2014) 168:229–33. doi: 10.1016/j.vetmic.2013.10.017
Resumen de PubMed | Texto completo de CrossRef | Google Académico
52. Possebon, FS, Tiba Casas, MR, Nero, LA, Yamatogi, RS, Araújo, JP Jr, y De AN, PJP. Prevalencia, resistencia a antibióticos, PFGE y caracterización MLST de Salmonella en ganglios linfáticos mesentéricos porcinos. Prev Vet Med. (2020) 179:105024. doi: 10.1016/j.prevetmed.2020.105024
Resumen de PubMed | Texto completo de CrossRef | Google Académico
53. Arfken, AM, Frey, JF, and Summers, KL. Temporal dynamics of the gut bacteriome and mycobiome in the weanling pig. Microorganismos. (2020) 8:868. doi: 10.3390/microorganismos8060868
Resumen de PubMed | Texto completo de CrossRef | Google Académico
54. Summers, KL, Frey, JF, Ramsay, TG y Arfken, AM. El micobioma de lechones durante la transición al destete: un estudio piloto. J Anim Sci. (2019) 97:2889–900. doi: 10.1093/jas/skz182
Resumen de PubMed | Texto completo de CrossRef | Google Académico
55. Patterson, SK, Kim, HB, Borewicz, K, e Isaacson, RE. Hacia una comprensión de la persistencia de Typhimurium por Salmonella enterica serovar en cerdos. Anim Health Res Rev. (2016) 17:159–68. doi: 10.1017/S1466252316000165
Resumen de PubMed | Texto completo de CrossRef | Google Académico
56. Argüello, H, Estellé, J, Leonard, FC, Crispie, F, Cotter, PD, O’Sullivan, O, et al. Influencia de la microbiota intestinal en la resistencia a la colonización de Salmonella y el patrón de desprendimiento de cerdos expuestos naturalmente. mSystems. (2019) 4:E00021–19. doi: 10.1128/mSystems.00021-19
57. Rivera-Chávez, F, López, CA, y Bäumler, AJ. El oxígeno como impulsor de la disbiosis intestinal. Libre Radic Biol Med. (2017) 105:93–101. doi: 10.1016/j.freeradbiomed.2016.09.022
Resumen de PubMed | Texto completo de CrossRef | Google Académico
58. Rivera-Chávez, F, Zhang, LF, Faber, F, Lopez, CA, Byndloss, MX, Olsan, EE, et al. El agotamiento de los clostridios productores de butirato de la microbiota intestinal impulsa una expansión luminal aeróbica de Salmonella. Microbio huésped celular. (2016) 19:443–54. doi: 10.1016/j.chom.2016.03.004
Resumen de PubMed | Texto completo de CrossRef | Google Académico
59. Litvak, Y, Mon, KKZ, Nguyen, H, Chanthavixay, G, Liou, M, Velazquez, EM, et al. Commensal Enterobacteriaceae protect against Salmonella colonization through oxygen competition. Microbio huésped celular. (2019) 25:128–139.e5. doi: 10.1016/j.chom.2018.12.003
Resumen de PubMed | Texto completo de CrossRef | Google Académico
60. Bukin, Y, Galachyants, Y, Morozov, I, Bukin, SV, Zakharenko, AS, y Zemskaya, TI. El efecto de la elección de la región 16S rRNA en los resultados de metabarcoding de la comunidad bacteriana. Datos Sci. (2019) 6:190007. doi: 10.1038/sdata.2019.7
Resumen de PubMed | Texto completo de CrossRef | Google Académico
61. Wang, X, Howe, S, Wei, X, Deng, F, Tsai, T, Chai, J, et al. El cultivo integral del microbioma intestinal porcino revela una alta diversidad bacteriana y guía el aislamiento bacteriano en cerdos. mSystems. (2021) 6:E00477–21. doi: 10.1128/mSystems.00477-21
62. Umu, ÖCO, Frank, JA, Fangel, JU, Oostindjer, M, da Silva, CS, Bolhuis, EJ, et al. La dieta de almidón resistente induce un cambio en el microbioma porcino y un predominio de poblaciones bacterianas beneficiosas. Microbioma. (2015) 3:16. DOI: 10.1186/S40168-015-0078-5
Resumen de PubMed | Texto completo de CrossRef | Google Académico
63. Tiwari, UP, Mandal, RK, Neupane, KR, Mishra, B, y Jha, R. Los piensos con almidón y fibrosos difieren en sus características de digestibilidad in vitro y fermentación y modulan de manera diferente la microbiota intestinal de los cerdos. J Anim Sci Biotechnol. (2022) 13:53. DOI: 10.1186/S40104-022-00699-Y
Resumen de PubMed | Texto completo de CrossRef | Google Académico
64. Helm, ET, Gabler, NK, y Burrough, ER. La fibra altamente fermentable altera la microbiota fecal y mitiga la disentería porcina inducida por Brachyspira hyodysenteriae. Animales. (2021) 11:396. doi: 10.3390/ani11020396
Resumen de PubMed | Texto completo de CrossRef | Google Académico
65. Liu, P, Zhao, J, Wang, W, Guo, P, Lu, W, Wang, C, et al. El salvado de maíz dietético alteró la diversidad de las comunidades microbianas y la producción de citoquinas en cerdos destetados. Microbiol frontal. (2018) 9:2090. DOI: 10.3389/fmicb.2018.02090
66. Oh, JK, Vasquez, R, Kim, SH, Hwang, I-C, Song, JH, Park, JH, et al. Los probióticos multiespecie alteran los ácidos grasos fecales de cadena corta y los niveles de lactato en cerdos destetados mediante la modulación de la microbiota intestinal. J Anim Sci Technol. (2021) 63:1142–58. DOI: 10.5187/JAST.2021.E94
Resumen de PubMed | Texto completo de CrossRef | Google Académico
67. Prabhu, R, Altman, E, y Eiteman, MA. Metabolismo de lactato y acrilato por Megasphaera elsdenii en condiciones de lote y estado estacionario. Aplicación Environ Microbiol. (2012) 78:8564–70. doi: 10.1128/AEM.02443-12
Resumen de PubMed | Texto completo de CrossRef | Google Académico
68. Gillis, CC, Winter, MG, Chanin, RB, Zhu, W, Spiga, L, and Winter, SE. Los metabolitos derivados del huésped modulan la transcripción de los genes de Salmonella implicados en la utilización de L-lactato durante la colonización intestinal. Infectar inmune. (2019) 87:E00773–18. doi: 10.1128/IAI.00773-18
Resumen de PubMed | Texto completo de CrossRef | Google Académico
69. Gillis, CC, Hughes, ER, Spiga, L, Winter, MG, Zhu, W, Furtado de Carvalho, T, et al. El cambio asociado a la disbiosis en el metabolismo del huésped genera lactato para apoyar el crecimiento de Salmonella. Microbio huésped celular. (2018) 23:54–64.e6. doi: 10.1016/j.chom.2017.11.006
Resumen de PubMed | Texto completo de CrossRef | Google Académico
70. Lauridsen, C. Effects of dietary fatty acids on gut health and function of pigs pre- and post-weaning. J Anim Sci. (2020) 98:skaa086. doi: 10.1093/jas/skaa086
Resumen de PubMed | Texto completo de CrossRef | Google Académico
71. Tett, A, Huang, KD, Asnicar, F, Fehlner-Peach, H, Pasolli, E, Karcher, N, et al. El complejo Prevotella copri comprende cuatro clados distintos subrepresentados en poblaciones occidentalizadas. Microbio huésped celular. (2019) 26:666–679.e7. doi: 10.1016/j.chom.2019.08.018
Resumen de PubMed | Texto completo de CrossRef | Google Académico
72. Fehlner-Peach, H, Magnabosco, C, Raghavan, V, Scher, JU, Tett, A, Cox, LM, et al. Distintos perfiles de utilización de polisacáridos de aislados de Prevotella copri intestinales humanos. Microbio huésped celular. (2019) 26:680–690.e5. doi: 10.1016/j.chom.2019.10.013
Resumen de PubMed | Texto completo de CrossRef | Google Académico
73. Nii, T, Maeda, Y, Motooka, D, Naito, M, Matsumoto, Y, Ogawa, T, et al. Repertorios genómicos vinculados con la potencia patógena de Prevotella copri artritogénica aislada del intestino de pacientes con artritis reumatoide. Ann Rheum Dis. (2023) 82:621–9. DOI: 10.1136/ARD-2022-222881
Resumen de PubMed | Texto completo de CrossRef | Google Académico
74. Arfken, AM, Frey, JF, Ramsay, TG, and Summers, KL. Yeasts of burden: exploring the mycobiome–bacteriome of the piglet GI tract. Microbiol frontal. (2019) 10:2286. doi: 10.3389/fmicb.2019.02286
Resumen de PubMed | Texto completo de CrossRef | Google Académico
75. Summers, KL, Foster Frey, J, y Arfken, AM. Caracterización de Kazachstania slooffiae, un comensal propuesto en el intestino porcino. J Hongos. (2021) 7:146. DOI: 10.3390/JUF7020146
Resumen de PubMed | Texto completo de CrossRef | Google Académico
76. Zhou, Z, Alikhan, N-F, Mohamed, K, Fan, Y, The Agama Study Group, y Achtman, M. La guía del usuario de EnteroBase, con estudios de casos sobre transmisiones de Salmonella, filogenia de Yersinia pestis y diversidad genómica central de Escherichia. Genoma Res. (2020) 30:138–52. DOI: 10.1101/GR.251678.119
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Palabras clave: Salmonella typhimurium, Salmonella 4,[5],12:i:- (monofásica), Prevotella, microbioma intestinal, porcino
Cita: Gomes-Neto JC, Pavlovikj N, Korth N, Naberhaus SA, Arruda B, Benson AK y Kreuder AJ (2023) Salmonella enterica induce cambios específicos de biogeografía en el microbioma intestinal de los cerdos. Frente. Vet. Sci. 10:1186554. doi: 10.3389/fvets.2023.1186554
Editado por:
Min Yue, Universidad de Zhejiang, China
Revisado por:
Rabindra Kumar Mandal, Universidad de Indiana, Universidad de Purdue Indianápolis, Estados Unidos
Thilo Fuchs, Instituto Friedrich-Loeffler, Alemania
Derechos de autor © 2023 Gomes-Neto, Pavlovikj, Korth, Naberhaus, Arruda, Benson y Kreuder. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia de Atribución Creative Commons (CC BY).
*Correspondencia: Amanda J. Kreuder, akreuder@iastate.edu
†Estos autores han contribuido igualmente a este trabajo
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o reclamo que pueda ser hecho por su fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía