
Diseño de una vacuna multiepítopo contra Haemophilus parasuis basada en enfoques pangenómicos e inmunoinformáticos

Diseño de una vacuna multiepítopo contra Haemophilus parasuis basada en enfoques pangenómicos e inmunoinformáticos
 Maonan Pang1,2†
Maonan Pang1,2†  Teng Tu1,2†
Teng Tu1,2†  Yin Wang1,2* Pengfei Zhang1,2 Meishen Ren1,2
Yin Wang1,2* Pengfei Zhang1,2 Meishen Ren1,2  Xueping Yao1,2 Yan Luo1,2 Zexiao Yang1,2
Xueping Yao1,2 Yan Luo1,2 Zexiao Yang1,2- 1Facultad de Medicina Veterinaria, Universidad Agrícola de Sichuan, Chengdu, China
- 2Laboratorio Clave de Enfermedades Animales y Salud Humana de la Provincia de Sichuan, Chengdu, Sichuan, China
Fondo: La enfermedad de Glässer, causada por Haemophilus parasuis (HPS), es responsable de pérdidas económicas en la industria porcina en todo el mundo. Sin embargo, las vacunas comerciales existentes ofrecen una protección deficiente y existen barreras significativas para el desarrollo de vacunas efectivas.
Métodos: En el presente estudio, nuestro objetivo era identificar posibles candidatos a vacunas y diseñar una vacuna multiepítopo contra el HPS mediante la realización de un análisis pangenómico de 121 cepas y el uso de un enfoque de vacunología inversa.
Resultados: Las construcciones de vacunas diseñadas consisten en epítopos predichos de células B y T derivados de las proteínas de la membrana externa del genoma central de HPS. Se descubrió que la vacuna es altamente inmunogénica, no tóxica y no alergénica, además de tener propiedades fisicoquímicas estables. Tiene una alta afinidad de unión al receptor tipo Toll 2. Además, los resultados de la simulación inmunitaria in silico mostraron que la vacuna provocó una respuesta inmunitaria eficaz. Además, el anticuerpo policlonal de ratón obtenido mediante la inmunización de la proteína de la vacuna puede combinarse con diferentes serotipos y Haemophilus parasuis in vitro no tipificables.
Conclusión: Los resultados generales del estudio sugieren que la vacuna multiepítopo diseñada es una candidata prometedora para la panprofilaxis contra diferentes cepas de HPS.
Introducción
La enfermedad causada por Haemophilus parasuis (HPS), conocida como enfermedad de Glässer, se caracteriza por poliserositis fibrinosa y artritis (Figuras 1, 2). Es una de las principales enfermedades infecciosas en el modelo de cría aislada de la industria porcina mundial y causa importantes pérdidas económicas (1). Las cepas de H. parasuis son heterogéneas en cuanto a rasgos fenotípicos y genotípicos. Las cepas se han clasificado en 15 serotipos, pero una gran proporción de aislados siguen siendo no tipificables (2). Actualmente, la vacunación es la principal medida para prevenir la infección por HPS. Las vacunas de bacterinas inactivadas disponibles en el mercado se basan en serovar 5, una combinación de serovares 4 y 5, o una combinación de serovares 1 y 6. Sin embargo, todos estos productos vacunales mostraron una protección cruzada limitada contra cepas heterólogas. A menudo incluso no se logra el efecto deseado en la protección contra diferentes aislamientos del mismo serotipo (3). Además, la protección contra las cepas no tipificables sigue siendo difícil de alcanzar. Además, más de una cepa de HPS suele estar presente en una granja porcina. Por ejemplo, se pueden aislar 4-5 cepas de un rebaño en un momento dado, y se pueden aislar hasta 16 cepas diferentes en una sola granja porcina durante un ciclo de producción (4-6). Esta característica epidemiológica también plantea un gran desafío para la selección de vacunas HPS en el proceso de mejoramiento.
 Figura 1. Grupo de cerdos de destete diagnosticados con enfermedad de Glässer. (A,B) Los cerdos se reúnen en la esquina del corral para protegerse del frío, sus cuerpos están sucios y sus pelajes están harapientos. Fotografías tomadas por el autor.
Figura 1. Grupo de cerdos de destete diagnosticados con enfermedad de Glässer. (A,B) Los cerdos se reúnen en la esquina del corral para protegerse del frío, sus cuerpos están sucios y sus pelajes están harapientos. Fotografías tomadas por el autor.
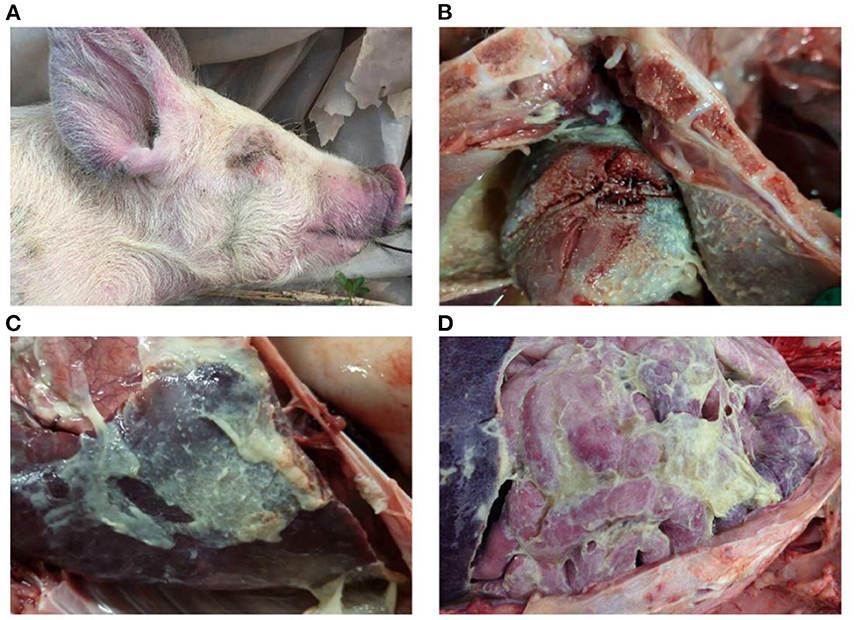 Figura 2. Lesiones macroscópicas de la enfermedad de Glässer: (A) marcas púrpuras en las orejas, la piel alrededor de los ojos y la punta de la nariz de un cerdo que murió de infección por H. parasuis; (B) exudado fibrinopurulento en la superficie pericárdica; y (C,D) exudado fibrinopurulento en membranas serosas en cavidades peritoneales y torácicas. Fotografías tomadas por el autor.
Figura 2. Lesiones macroscópicas de la enfermedad de Glässer: (A) marcas púrpuras en las orejas, la piel alrededor de los ojos y la punta de la nariz de un cerdo que murió de infección por H. parasuis; (B) exudado fibrinopurulento en la superficie pericárdica; y (C,D) exudado fibrinopurulento en membranas serosas en cavidades peritoneales y torácicas. Fotografías tomadas por el autor.
Dados los retos a los que se enfrentan las vacunas bacterianas inactivadas para el tratamiento del SPH descritos anteriormente, el uso de la vacunología inversa para desarrollar vacunas proteicas contra los epítopos protectores del patógeno es una estrategia viable. La vacunología inversa implica programas informáticos para identificar epítopos antigénicos basados en la información de la secuencia del genoma bacteriano para el desarrollo y diseño de vacunas, evitando las desventajas del diseño tradicional de vacunas, que es costoso y requiere mucho tiempo (7, 8). Además, con la constante actualización de las tecnologías de secuenciación, se puede obtener información de secuencia de genomas bacterianos a bajo costo y en poco tiempo, lo que también reduce el tiempo requerido para el diseño de vacunas.
Por lo tanto, en este estudio, utilizamos el análisis del genoma pan para identificar el genoma central de HPS. Luego, se realizó una predicción in silico de los epítopos de las células B y T de las proteínas de la membrana externa en el genoma central para diseñar una vacuna multiepítopo. También se ligó un adyuvante a la vacuna para mejorar la inmunogenicidad de la vacuna y obtener la construcción final de la vacuna multiepítopo. Posteriormente, se estimó la antigenicidad y las propiedades fisicoquímicas de la construcción de la vacuna. Además, se predijeron las estructuras secundaria y terciaria del constructo y se evaluó la interacción de la vacuna con el receptor tipo Toll 2 mediante simulaciones de acoplamiento molecular. Finalmente, se realizaron simulaciones inmunes para confirmar el potencial inmune de la construcción de la vacuna y se construyó un vector para su expresión en E. coli. El potencial de aplicación de la vacuna multiepítopo se probó preliminarmente en la prueba de inmunización con ratones. Así, en este trabajo, se creó un candidato vacunal multiepítopo utilizando una novedosa estrategia de diseño de vacunas basada en el análisis pangenómico y técnicas de vacunología inversa que también ayudará a acelerar el desarrollo de vacunas contra otros patógenos.
Materiales y métodos
Cepas bacterianas
En este estudio, recuperamos el genoma completo de 105 cepas de HPS con una rica diversidad geográfica, virulenta y serológica, que estaban disponibles en marzo de 2020 en el NCBI (ftp://ftp.ncbi.nih.gov/genomes/all/). La información sobre las 105 cepas se resume en la Tabla Suplementaria 1.
Además, se secuenciaron 16 cepas clínicas aisladas en Sichuan entre 2015 y 2020. El ADN se extrajo del cultivo nocturno utilizando un kit de ADN bacteriano E.Z.N.A (OMEGA) siguiendo las directrices del fabricante y se secuenció utilizando la plataforma Min-ION MK1B. Las lecturas de ONT sin procesar se corrigieron utilizando Canu (9) (v1.5 https://github.com/marbl/canu, consultado el 15 de octubre de 2020) y luego se utilizó SMARTdenovo (https://github.com/ruanjue/smartdenovo, consultado el 15 de octubre de 2020) para ensamblar las lecturas corregidas por errores para obtener la secuencia del genoma ensamblado. Por último, se utilizaron Medaka (v1.0.1https://github.com/nanoporetech/medaka, consultado el 15 de octubre de 2020) y Homopolish (10) (v0.2.1 https://github.com/ythuang0522/homopolish, consultado el 15 de mayo de 2021) para realizar varias rondas de pulido de ensamblajes de desmoldeo. Se aislaron ciento quince cepas de HPS de casos clínicos con enfermedad de Glässer, y seis cepas fueron del tracto respiratorio superior de cerdos sanos.
Análisis pangenómico y filogenético
Para mantener la coherencia y la fiabilidad de la predicción y la anotación de genes, se aplicó uniformemente a los 121 genomas de HPS una herramienta de software estándar, la canalización del Sistema de Anotación del Genoma Procariota (Prokka) (11) (v1.14.5 https://github.com/tseemann/prokka, consultada el 5 de septiembre de 2021). Sobre la base de los archivos GFF3 producidos por Prokka, se utilizó el programa Roary (12) (https://github.com/sanger-pathogens/Roary, consultado el 6 de octubre de 2021) para llevar a cabo el análisis pangenómico con el fin de identificar genes centrales y accesorios con un porcentaje mínimo de identidad del 95% entre cada proteína homóloga predicha. A continuación, se construyó un árbol NJ (neighbor-joining) de acuerdo con los genes centrales de las cepas HPS utilizando MEGA (13) con 1.000 replicaciones bootstrap.
Selección de secuencias proteicas para el diseño de vacunas
Las secuencias proteicas de los genes centrales y los genes blandos se extrajeron en función de los resultados del análisis pangenómico. A continuación, se utilizó el servidor SignalP 5.0 (14) para analizar la presencia de péptidos señal (https://services.healthtech.dtu.dk/service.php?SignalP-5.0, consultado el 3 de noviembre de 2021) y diferenciar entre proteínas secretoras y no secretoras. La localización subcelular de las proteínas secretadas se comprobó además en Vaxign (http://www.violinet.org/vaxign/, consultado el 3 de noviembre de 2021) para seleccionar proteínas de membrana externa como candidatas para la construcción de vacunas (15).
Predicción de epítopos de células B
Las células B son un componente central del sistema inmunitario adaptativo y proporcionan protección a largo plazo contra patógenos infecciosos mediante la producción de anticuerpos. En este estudio, los epítopos lineales de células B de las proteínas candidatas fueron predichos por el servidor web BepiPred-2.0 (16) (https://services.healthtech.dtu.dk/service.php?BepiPred-2.0, consultado el 4 de noviembre de 2021).
Predicción de epítopos de linfocitos T citotóxicos (CTL) y linfocitos T colaboradores (HTL)
Los epítopos de linfocitos T (CTL) citotóxicos a partir de secuencias de proteínas candidatas se predijeron utilizando el servidor NetCTL 1.2 (https://services.healthtech.dtu.dk/service.php?NetCTL-1.2, consultado el 4 de noviembre de 2021). Se utilizó la configuración predeterminada (umbral, 0,75) para la estimación de los epítopos CTL (17). A continuación, se predijeron los epítopos de 15 meros de las células T auxiliares para las secuencias de proteínas candidatas utilizando el servidor NetMHCII 2.3 (https://services.healthtech.dtu.dk/service.php?NetMHCII-2.3, consultado el 4 de noviembre de 2021). Se evaluaron siete alelos H2 de ratón clase II. De acuerdo con los estándares, las puntuaciones de consenso más bajas de los péptidos se eligieron como los mejores aglutinantes y un rango de percentil más bajo indica una mayor afinidad. El criterio de selección fue un punto de corte de IC50 ≤ 50 y rango percentil <1 (18).
Construcción de vacunas
Se diseñó una supuesta secuencia candidata a vacuna combinando epítopos de células B con epítopos CTL de alta puntuación y epítopos HTL de alta afinidad de unión. El agonista de TLR-2, proteína modulina α4 soluble en fenol (n.º de acceso A9JX08), fue preferido como adyuvante para mejorar la inmunogenicidad de la vacuna (19). El adyuvante se unió al primer epítopo de la célula B a través de un enlazador EAAAK en el terminal N de la secuencia, mientras que los epítopos restantes de la célula B y la HTL se interconectaron a través de enlazadores GPGPG. Se utilizaron enlazadores AAY para unir los epítopos de las CTL.
Evaluación de la antigenicidad, alergenicidad y propiedades fisicoquímicas de la proteína
Para predecir la antigenicidad y la alergenicidad de la vacuna, se utilizaron el servidor VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html, consultado el 5 de noviembre de 2021) y el servidor AllerTOP v2.0 (http://www.ddg-pharmfac.net/AllerTOP, consultado el 5 de noviembre de 2021), respectivamente (20, 21). La solubilidad de la vacuna diseñada se evaluó utilizando el servidor SOLpro (22) (https://scratch.proteomics.ics.uci.edu, consultado el 5 de noviembre de 2021). Además, se evaluaron varias propiedades fisicoquímicas de la vacuna diseñada mediante el servidor ProtParam (http://web.expasy.org/protparam/, consultado el 5 de noviembre de 2021).
Extrapolación de la estructura secundaria de la proteína
La estructura secundaria de la vacuna multiepítopo se predijo utilizando el servidor PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/, consultado el 6 de noviembre de 2021) y RaptorX (http://raptorx.uchicago.edu/StructurePropertyPred/predict/, consultado el 6 de noviembre de 2021) con parámetros predeterminados (23, 24).
Modelado tridimensional y validación de la proteína
El modelo de homología de la construcción final de la vacuna se realizó utilizando el servidor Robetta (25) (https://robetta.bakerlab.org/, consultado el 7 de noviembre de 2021). Las interacciones no enlazantes entre diferentes tipos de átomos se analizaron utilizando el servidor ERRAT (26) (http://services.mbi.ucla.edu/ERRAT/, consultado el 7 de noviembre de 2021) para verificar la estructura terciaria. El diagrama de Ramachandran se generó utilizando el servidor PROCHECK (27) (https://servicesn.mbi.ucla.edu/PROCHECK/, consultado el 7 de noviembre de 2021) para determinar la proporción relativa de aminoácidos en las regiones favorecidas.
Predicción de epítopos discontinuos de células B
ElliPro (http://tools.iedb.org/ellipro/, consultado el 7 de noviembre de 2021) se utilizó para la predicción de epítopos discontinuos de células B en el modelo de proteína (28).
Acoplamiento molecular de la proteína con TLR2
Los receptores tipo Toll son sensores de la respuesta inmunitaria innata, y TLR-2 reconoce la gama más amplia de moléculas PAMPs e induce respuestas antibacterianas y antivirales significativas. Utilizamos el servidor ClusPro (https://cluspro.bu.edu/login.php, consultado el 8 de noviembre de 2021) para determinar el acoplamiento de TLR-2 con la proteína de la vacuna (29). Las coordenadas estructurales del TLR-2 (PDB ID: 3A7C) se obtuvieron del Banco de Datos de Proteínas (30) (https://www.rcsb.org/, consultado el 8 de noviembre de 2021). Las estructuras acopladas se visualizaron a través de PyMOL (http://www.pymol.org, consultado el 8 de noviembre de 2021) para analizar la interacción entre la proteína de la vacuna y TLR-2.
Caracterización del perfil inmune de constructo
Para el análisis de las respuestas inmunitarias de la construcción de la vacuna en el modelo de ratón, se empleó el servidor de simulación inmunitaria dinámica en línea C-lmmSim (https://www.iac.cnr.it/~filippo/c-immsim/index.html, consultado el 9 de noviembre de 2021) (31). Este servidor en línea funciona en base a una matriz de puntuación específica de posición (PSSM) para la predicción de epítopos inmunogénicos e interacciones inmunitarias. Se utilizaron todos los parámetros de simulación predeterminados con periodos de tiempo especificados en 1, 90 y 180.
Optimización in silico y clonación de la proteína
Para garantizar la expresión eficiente de la construcción de la vacuna en células de Escherichia coli, realizamos la traducción inversa y la optimización de codones de la secuencia de proteínas de la vacuna utilizando la herramienta Java Codon Adaptation Tool (32) (http://www.jcat.de/, consultado el 9 de noviembre de 2021). Se seleccionó la cepa K12 de E. coli como huésped de expresión. Durante la ejecución, las opciones se eligieron para evitar los terminadores de transcripción independientes de rho, los sitios de unión de ribosomas bacterianos y los sitios de escisión de enzimas de restricción. Por último, la secuencia de la proteína de la vacuna se diseñó para su clonación en un vector huésped adecuado, pET-28a(+)-MEV, empleando el software SnapGene (https://www.snapgene.com/, consultado el 9 de noviembre de 2021).
Expresión inducible y purificación de proteínas vacunales
El vector de expresión sintética llamado pET-28a(+)-MEV está disponible en Chengdu YouKang Jianxing Biotechnology Co. El pET-28a(+)-MEV se transformó en E. coli BL21 (DE3), y las colonias positivas se inocularon en medio líquido LB que contenía kanamicina y se incubaron durante la noche en un agitador de baño de agua a 37°C. Se inoculó una alícuota de 10 mL de cultivo durante la noche en 1 L de medio LB que contenía kanamicina, y el cultivo se expandió a crecimiento logarítmico medio (OD600 ≈ 0,6) a 37 °C en un agitador en baño de agua. Se añadió IPTG al cultivo y se incubó a 37°C durante 5 h. Las bacterias se recogieron por centrifugación y se fragmentaron por ultrasonidos. Después de la sonicación, la proteína de la vacuna se purificó utilizando un Ni2+-Columna de resina NTA y concentrada mediante desalinización mediante un filtro centrífugo Amicon Ultra-4 10K.®
Inmunoensayo en ratón y ensayo ELISA
Diez ratones Kunming de 6 semanas de edad de grado SPFKM se dividieron aleatoriamente en grupos de prueba y control de cinco ratones cada uno. La proteína de la vacuna se diluyó a 1 μg/μL y 50 μL se mezclaron con un volumen igual de inmunoadyuvante de anticuerpos rápidos. Se inyectó por vía intramuscular una alícuota de 100 μL de la solución vacunal en las patas traseras de ratones, que fueron inmunizados de nuevo con la misma dosis el día 14. Al grupo control se le inyectó una mezcla de solución salina y adyuvante inmunológico. Se extrajo sangre de la vena de la cola de los ratones a los 21 días después de la inmunización y se aisló y conservó el suero. Dieciséis aislamientos clínicos de HPS se recubrieron en placas de ELISA y se midió la absorbancia a OD450 y se registraron los datos. El análisis estadístico y la visualización de los datos se realizaron utilizando R 4.1.
Resultados
Secuenciación de nanoporos y ensamblaje del genoma de 16 aislamientos clínicos de HPS
Para construir un pangenoma más completo de HPS, se secuenciaron 16 aislamientos clínicos de HPS (HPS-1-HPS-15) utilizando la plataforma ONT MinION MK1B. Se obtuvieron lecturas de cada cepa con una profundidad de aproximadamente 100 ×. Las lecturas de ONT sin procesar se corrigieron con Canu, y luego se utilizó SMARTdenovo para ensamblar las lecturas corregidas por errores para obtener la secuencia del genoma ensamblado. A continuación, las lecturas en bruto se sometieron a múltiples rondas de pulido utilizando Medaka y Homopolish para mejorar la calidad de los conjuntos de borrador. Los datos que respaldan los hallazgos de este estudio han sido depositados en el CNGB Sequence Archive (CNSA) de la Base de Datos del Banco Nacional de Germoplasma de China (CNGBdb) con número de acceso CNP0002150 (33, 34). La información sobre las 16 cepas se resume en la Tabla Suplementaria 2.
Resultados del análisis pangenómico y filogenético
El pangenoma observado compartido por las 121 cepas HPS consta de 8.885 genes, incluidos 390 genes principales y 8.495 genes accesorios (Figura 3A). El análisis de regresión no lineal mostró un pangenoma abierto evidente, y el tamaño de los genes centrales se acercó a un valor constante (Figura 3B). Sobre la base de genes centrales concatenados, construimos un árbol filogenético del HPS, que mostró la rica diversidad filogenética del HPS (Figura 3C). Estos resultados indican que el genoma del HPS es muy variable y que puede obtener continuamente genes extraños para adaptarse a diferentes ambientes. Esta es la razón principal por la que las vacunas convencionales son menos protectoras.
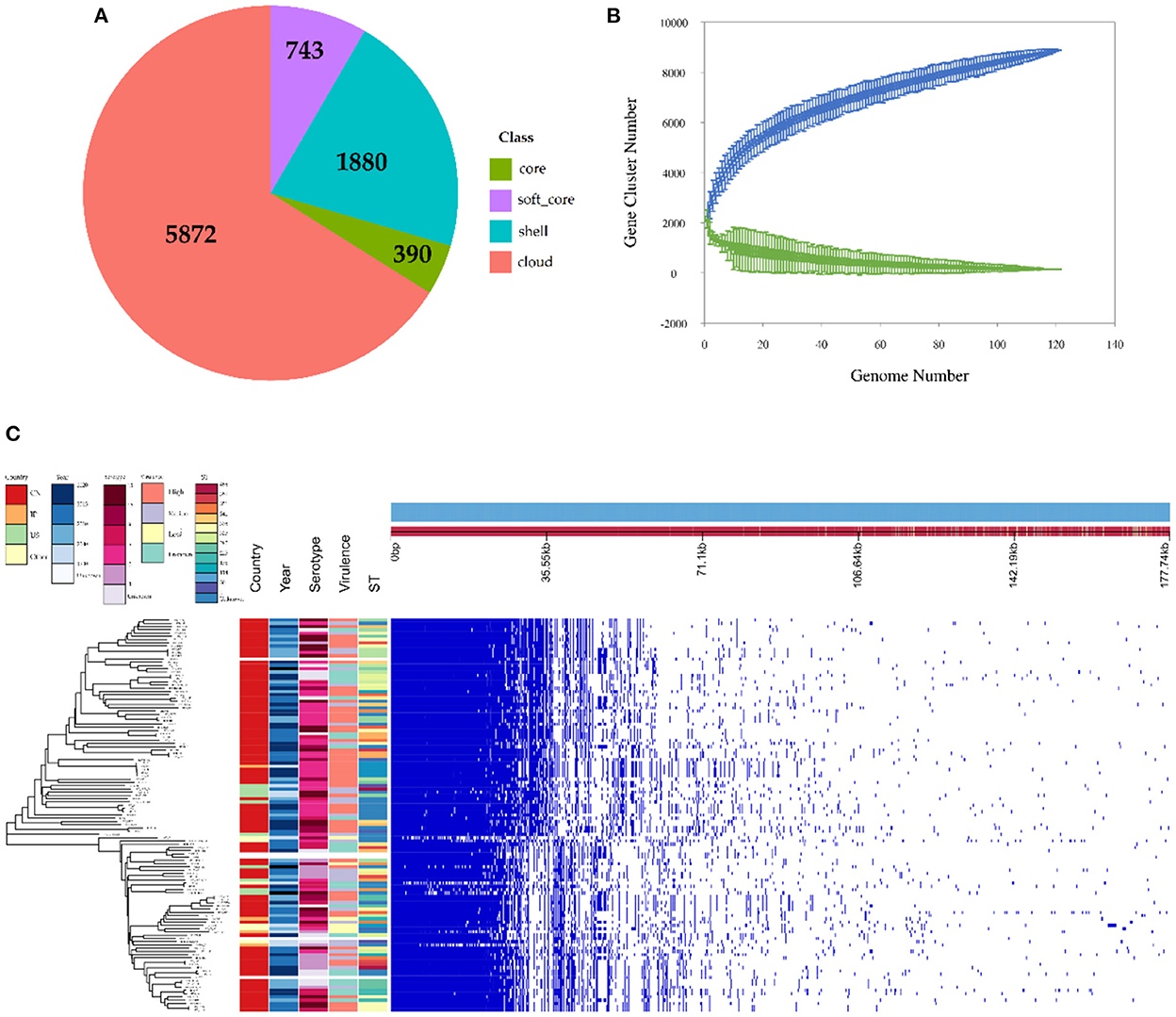 Figura 3. Análisis pangenómico y filogenético de HPS basado en 121 cepas. (A) Genes centrales y accesorios de HPS. Núcleo: 119 cepas ≤ ≤ 121; núcleo blando: 114≤ cepas < 119; concha: 18 cepas ≤ < 114; Nube: cepas < 18. (B) Curvas del núcleo y del pangenoma para HPS. Los puntos y líneas azules indican el tamaño del pangenoma para cada combinación de cepas, y la relación entre el tamaño del pangenoma y el número de genoma, respectivamente. Los puntos y líneas verdes indican la información correspondiente al genoma central. (C) El árbol de la izquierda es un árbol filogenético de unión de vecinos construido en base a los genes centrales y anotado con la ubicación del aislamiento, el tiempo de aislamiento, el serotipo, el tipo ST y la virulencia de cada cepa. El gráfico de la matriz de la derecha denota la presencia y ausencia de cada gen en todas las cepas por azul y blanco, respectivamente.
Figura 3. Análisis pangenómico y filogenético de HPS basado en 121 cepas. (A) Genes centrales y accesorios de HPS. Núcleo: 119 cepas ≤ ≤ 121; núcleo blando: 114≤ cepas < 119; concha: 18 cepas ≤ < 114; Nube: cepas < 18. (B) Curvas del núcleo y del pangenoma para HPS. Los puntos y líneas azules indican el tamaño del pangenoma para cada combinación de cepas, y la relación entre el tamaño del pangenoma y el número de genoma, respectivamente. Los puntos y líneas verdes indican la información correspondiente al genoma central. (C) El árbol de la izquierda es un árbol filogenético de unión de vecinos construido en base a los genes centrales y anotado con la ubicación del aislamiento, el tiempo de aislamiento, el serotipo, el tipo ST y la virulencia de cada cepa. El gráfico de la matriz de la derecha denota la presencia y ausencia de cada gen en todas las cepas por azul y blanco, respectivamente.
Secuencias de proteínas para el diseño de vacunas
Se seleccionaron ocho proteínas con péptidos señal secretados localizados en la membrana externa de los genes central y soft-core utilizando Signal 5.0 y Vaxign (Tabla 1). Estas secuencias de proteínas se sometieron a la predicción de epítopos en células B, T y auxiliares.
 Tabla 1. Proteínas seleccionadas en los genes core y soft-core de HPS.
Tabla 1. Proteínas seleccionadas en los genes core y soft-core de HPS.
Predicción de epítopos de linfocitos B, linfocitos T citotóxicos (CTL) y linfocitos T colaboradores (HTL)
Se utilizó el servidor BepiPred 2.0 para seleccionar epítopos de células B con el umbral predeterminado > 0,6. Los epítopos que están expuestos y tienen una estructura enrollada se seleccionaron para el diseño de la vacuna (Tabla 2). Las secuencias de proteínas fueron analizadas por el servidor NetCTL 1.2 para identificar las regiones más inmunodominantes. Los péptidos con las puntuaciones más altas de afinidad de unión en cada proteína se identificaron como candidatos a epítopos CTL de alto potencial y se examinaron un total de tres epítopos (Tabla 2). El servidor web NetMHCII 2.3 predijo los epítopos MHC-II con la mayor unión correspondiente a los alelos en función de la puntuación IC50. Se eligieron un total de 6 epítopos HTL para la construcción quimérica final (Tabla 2).
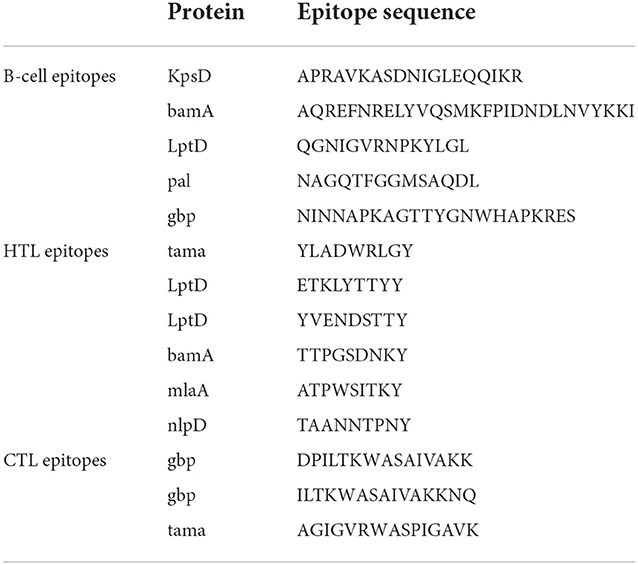 Tabla 2. Epítopos predichos de células B, CTL y HTL para el diseño de la proteína de la vacuna.
Tabla 2. Epítopos predichos de células B, CTL y HTL para el diseño de la proteína de la vacuna.
Construcción de vacunas
Se utilizaron un total de 5 epítopos de células B, seis epítopos HTL y tres epítopos HTL para construir quimeras de vacunas multiepítopos. Los epítopos de células B y HTL se unieron entre sí mediante enlazadores GPGPG, mientras que los epítopos AY se utilizaron para unir los epítopos CTL. Se añadió modulina soluble fenólica α4 (UniProt Id: A9JX08), un agonista de TLR-2, como adyuvante en el extremo N-terminal con un enlazador EAAAK para mejorar la inmunogenicidad de la vacuna. La construcción quimérica final constituyó 280 aminoácidos (Figura 4).
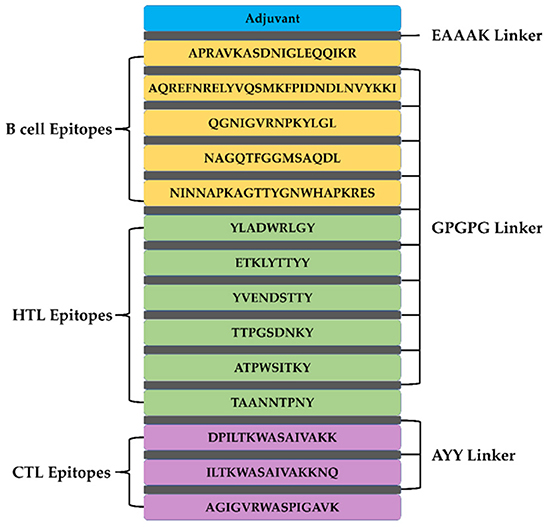 Figura 4. La disposición estructural de los epítopos de las células B y T junto con los enlazadores y el adyuvante para la construcción final de la vacuna multiepítopo.
Figura 4. La disposición estructural de los epítopos de las células B y T junto con los enlazadores y el adyuvante para la construcción final de la vacuna multiepítopo.
Antigenicidad, alergenicidad y propiedades fisicoquímicas de la proteína de la vacuna
Se utilizó el servidor web VaxiJen 2.0 para predecir la antigenicidad del diseño de la vacuna adjunto a un adyuvante, como 0,7756 con el modelo bacteriano optando por un umbral de 0,4. También se comprobó la antigenicidad de la vacuna candidata sin incluir la parte adyuvante para la que VaxiJen dio puntuaciones de 0,7114 en un modelo de bacterias. Los servidores en línea de AllerTOP v.2 y AllergenFP predijeron que la secuencia de la vacuna no sería de naturaleza alergénica en presencia y ausencia del adyuvante.
Solubilidad y propiedades fisicoquímicas de la proteína de la vacuna
El servidor SOLpro de la herramienta de predicción de proteínas Scratch predijo una probabilidad de solubilidad de 0,887834 para esta proteína de la vacuna. Se utilizó ExPASy ProtParam para predecir el peso molecular (MW) de la proteína de la vacuna como 29,4 kDa. El pI (valor teórico del punto isoeléctrico) de la proteína se calculó como 9,65. La vida media estimada de la proteína en mamíferos, levaduras y E. coli se estimó en 30 h, >20 h y >10 h, respectivamente. Además, ProtParam predijo que la proteína tenía un índice de inestabilidad (II) de 24,99, clasificándola como estable ya que un valor de >40 indica inestabilidad. Los valores de 70,03 y −0,438 para el índice alifático y GRAVY (gran promedio de hidropaticidad) reflejan la alta termoestabilidad y la naturaleza hidrofílica, respectivamente.
Estructura secundaria de la proteína de la vacuna
Sobre la base de los resultados de los servidores PSIPRED y RaptorX, la vacuna proteica consta de un 22 % de elementos estructurales secundarios de hélice (H), 3 % de hebra beta (E) y 73 % de espiral (C) (Figura 5A). Sobre la base de la accesibilidad de los residuos de aminoácidos, se predijo que el 62% de los residuos estarían expuestos, el 18% al medio y el 18% al enterramiento (Figura 5B). En total, solo el 2% de los residuos se localizaron en dominios estructurales desordenados.
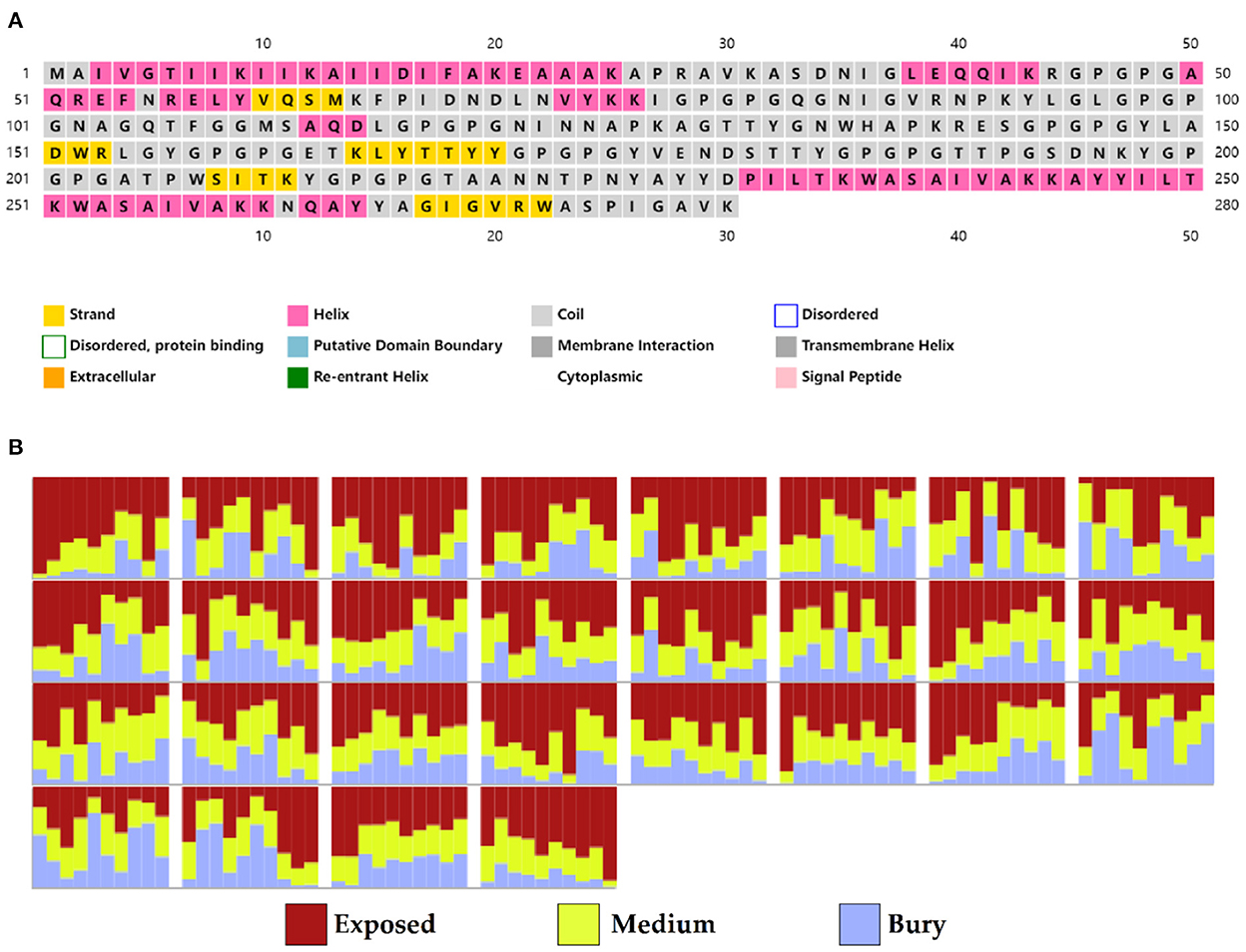 Figura 5. Predicción de la estructura secundaria de la secuencia de proteínas de la vacuna mediante el uso de PSIPRED y el servidor RaptorX que muestra (A) elementos estructurales secundarios y (B) accesibilidad al solvente según tres estados.
Figura 5. Predicción de la estructura secundaria de la secuencia de proteínas de la vacuna mediante el uso de PSIPRED y el servidor RaptorX que muestra (A) elementos estructurales secundarios y (B) accesibilidad al solvente según tres estados.
Modelado tridimensional y validación de la proteína de la vacuna
El modelo 3D de la proteína de la vacuna fue generado por el servidor Robetta (Figura 6) y se visualizó utilizando el software PyMOL. El análisis de la parcela de Ramachandran mostró que el 100% de los residuos se encontraban en las regiones permitidas y el 91,1% de los residuos en las regiones más favorecidas. La puntuación global del factor de calidad generado por el ERRAT2 fue del 92,647%. Los resultados anteriores para el modelo 3D demuestran que la construcción de la vacuna tiene una estructura química estable.
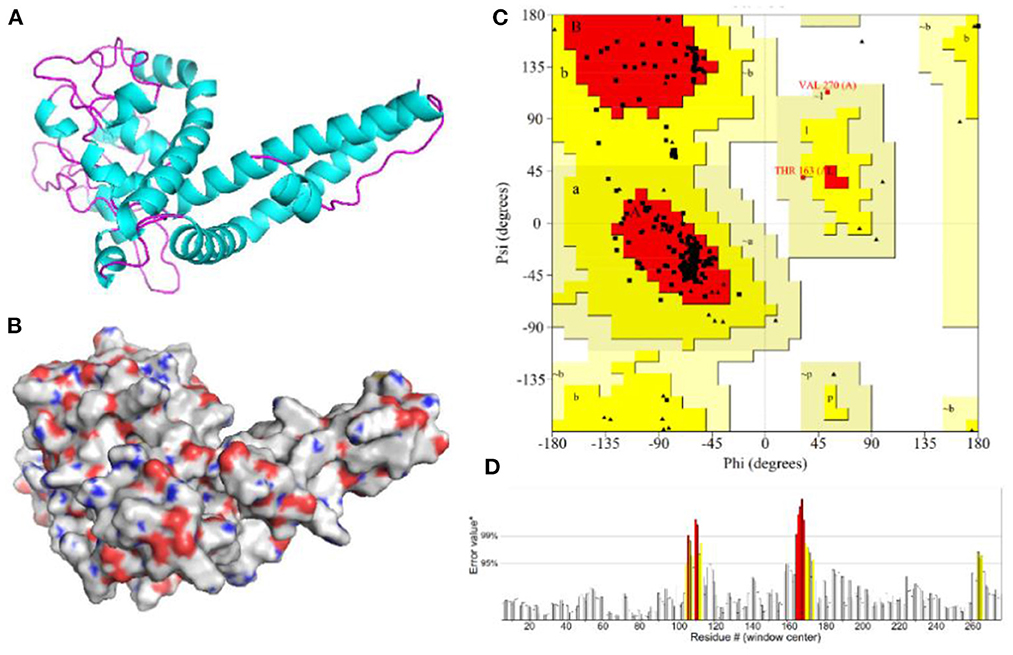 Figura 6. Modelización de homología y validación de la estructura tridimensional de construcciones de vacunas. (A,B) Modelos 3D de construcciones de vacunas generados por modelado de homología en Robetta y visualizados por el software PyMOL. (C) El análisis de la parcela de Ramachandran muestra el 100% de los residuos en la región permitida. (D) La puntuación global del factor de calidad del modelo fue del 92,647%.
Figura 6. Modelización de homología y validación de la estructura tridimensional de construcciones de vacunas. (A,B) Modelos 3D de construcciones de vacunas generados por modelado de homología en Robetta y visualizados por el software PyMOL. (C) El análisis de la parcela de Ramachandran muestra el 100% de los residuos en la región permitida. (D) La puntuación global del factor de calidad del modelo fue del 92,647%.
Predicción de epítopos discontinuos de células B
Se utilizó el servidor ElliPro para predecir epítopos discontinuos de células B para las construcciones de la vacuna. Los resultados mostraron que las construcciones de la vacuna tenían cinco epítopos discontinuos de células B que variaban en tamaño de 7 a 38 residuos, con puntuaciones que oscilaban entre 0,588 y 0,822 (Figura 7 y Tabla 3).
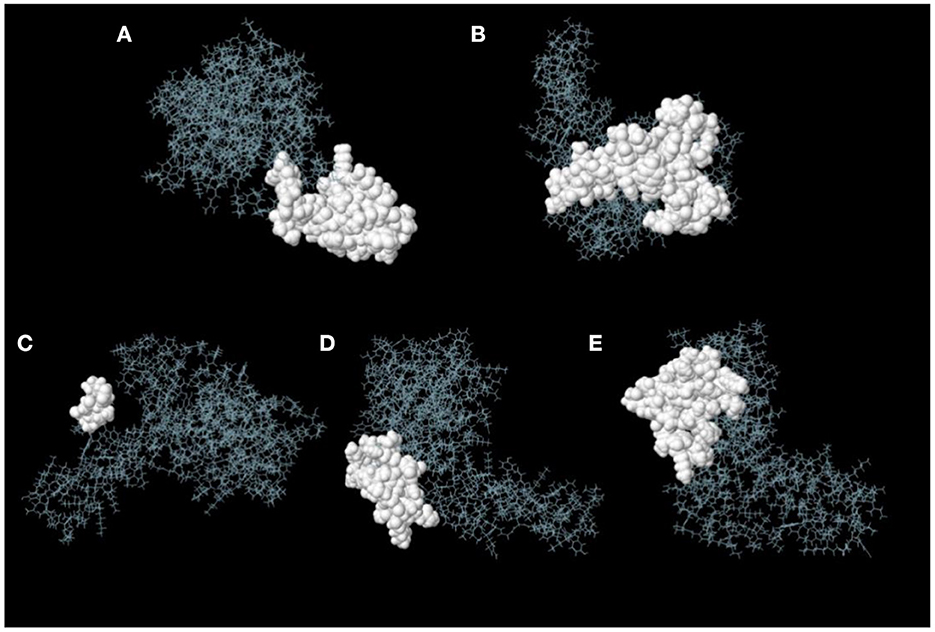 Figura 7. Epítopos discontinuos de células B de construcciones de vacunas predichas por ElliPro. (A-E) Los epítopos están representados por una superficie blanca. Las varillas grises corresponden al resto de la proteína.
Figura 7. Epítopos discontinuos de células B de construcciones de vacunas predichas por ElliPro. (A-E) Los epítopos están representados por una superficie blanca. Las varillas grises corresponden al resto de la proteína.
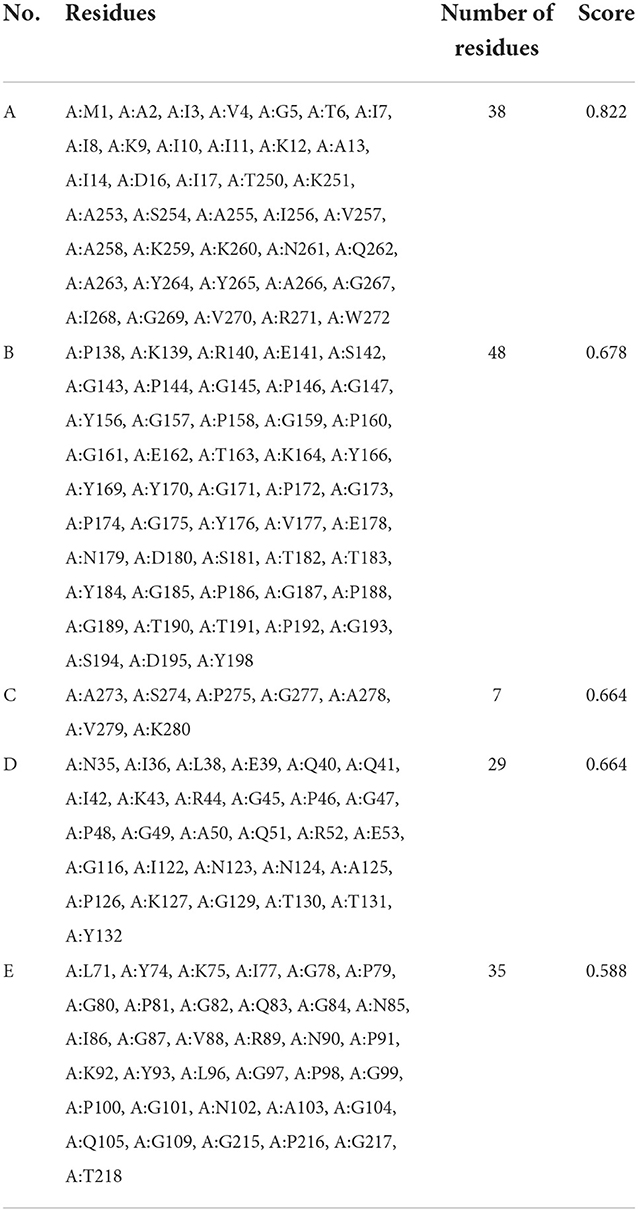 Tabla 3. Epítopos discontinuos de células B de construcciones de vacunas predichas por ElliPro.
Tabla 3. Epítopos discontinuos de células B de construcciones de vacunas predichas por ElliPro.
Acoplamiento molecular de la proteína de la vacuna con TLR2
Las células inmunitarias reconocen patrones de patógenos conservados evolutivamente de manera específica y responden expresando receptores similares a Toll. Dentro de la familia TLR, TLR2 tiene un amplio espectro de reconocimiento, reconociendo lipoproteínas, lipopolisacáridos, peptidoglicanos y otras señales que indican peligro (35). Por lo tanto, utilizamos el servidor ClusPro para determinar el acoplamiento de TLR 2 con la proteína de la vacuna. La energía central entre el receptor del ligando es −820.2, y la energía más baja del complejo de acoplamiento es −857.6. La proteína de la vacuna tiene múltiples interacciones enlazadas por hidrógeno con TLR2 y exhibe una alta afinidad de unión. Los residuos del complejo vacuna-TLR2 que muestran interacciones H-enlace son LYS25-ASP294, ARG28-GLY293, ARG28-PHE295, ARG271-LEU350, SER274-ASN379, ALA278-ASN379, LYS259-ARG400, LYS259-GLU374 y LYS259-GLU375 con una distancia de 2,1, 1,8, 1,8, 2,2, 2,4, 1,6, 2,1, 1,9 y 1,9Å, respectivamente (Figura 8).
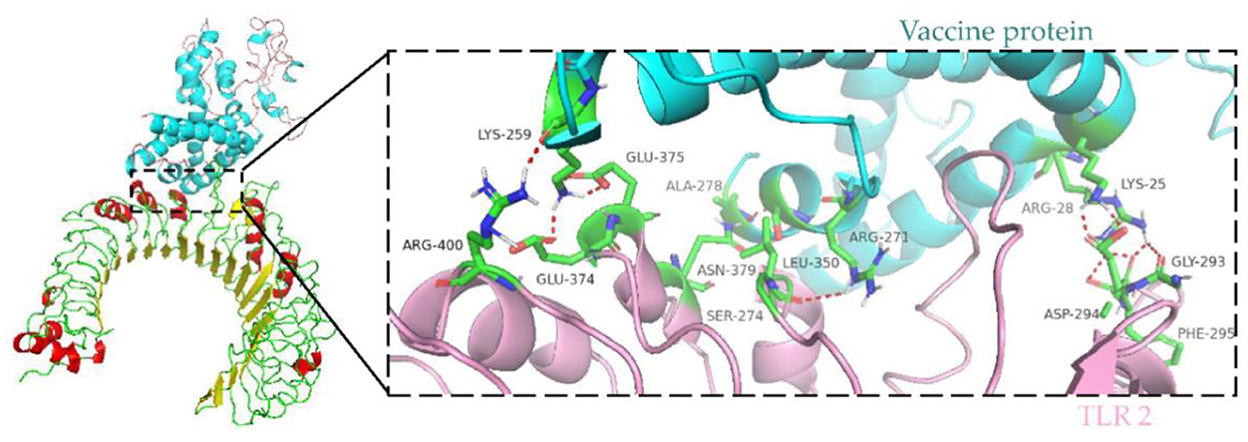 Figura 8. Acoplamiento molecular de la vacuna de subunidades con TLR2. Complejo de acoplamiento de la proteína de la vacuna y TLR2, con la proteína de la vacuna de color azul cielo y la cadena A de TLR2 de color rosa claro. Los residuos con interacciones de enlace H se representan en el modelo de palos y los residuos restantes se representan en el modelo de dibujos animados. Los enlaces de hidrógeno se representan como líneas discontinuas rojas.
Figura 8. Acoplamiento molecular de la vacuna de subunidades con TLR2. Complejo de acoplamiento de la proteína de la vacuna y TLR2, con la proteína de la vacuna de color azul cielo y la cadena A de TLR2 de color rosa claro. Los residuos con interacciones de enlace H se representan en el modelo de palos y los residuos restantes se representan en el modelo de dibujos animados. Los enlaces de hidrógeno se representan como líneas discontinuas rojas.
Simulación inmunitaria de la construcción de la vacuna
La caracterización inmunológica de la construcción de la vacuna diseñada se analizó utilizando el servidor C-ImmSim. Los resultados de la simulación inmunitaria del servidor C-ImmSim confirmaron la consistencia con las reacciones inmunitarias reales. Como se muestra en la Figura 9A, la producción de IgM se registró en la primera inyección de la vacuna y se observaron niveles elevados de IgM+IgG e IgG1+IgG2 después de la segunda y tercera inmunización. Se observaron altos niveles de poblaciones de células B después de múltiples inyecciones de las construcciones de la vacuna, lo que indica la formación de memoria inmunitaria (Figura 9C). Además, se observó un aumento de la citotoxicidad y de los linfocitos T colaboradores tras la vacunación, lo que indica la activación de respuestas inmunitarias mediadas por células (Figuras 9B, D). Los resultados anteriores demuestran la capacidad de la construcción de la vacuna para inducir una respuesta inmunitaria eficaz para eliminar el antígeno.
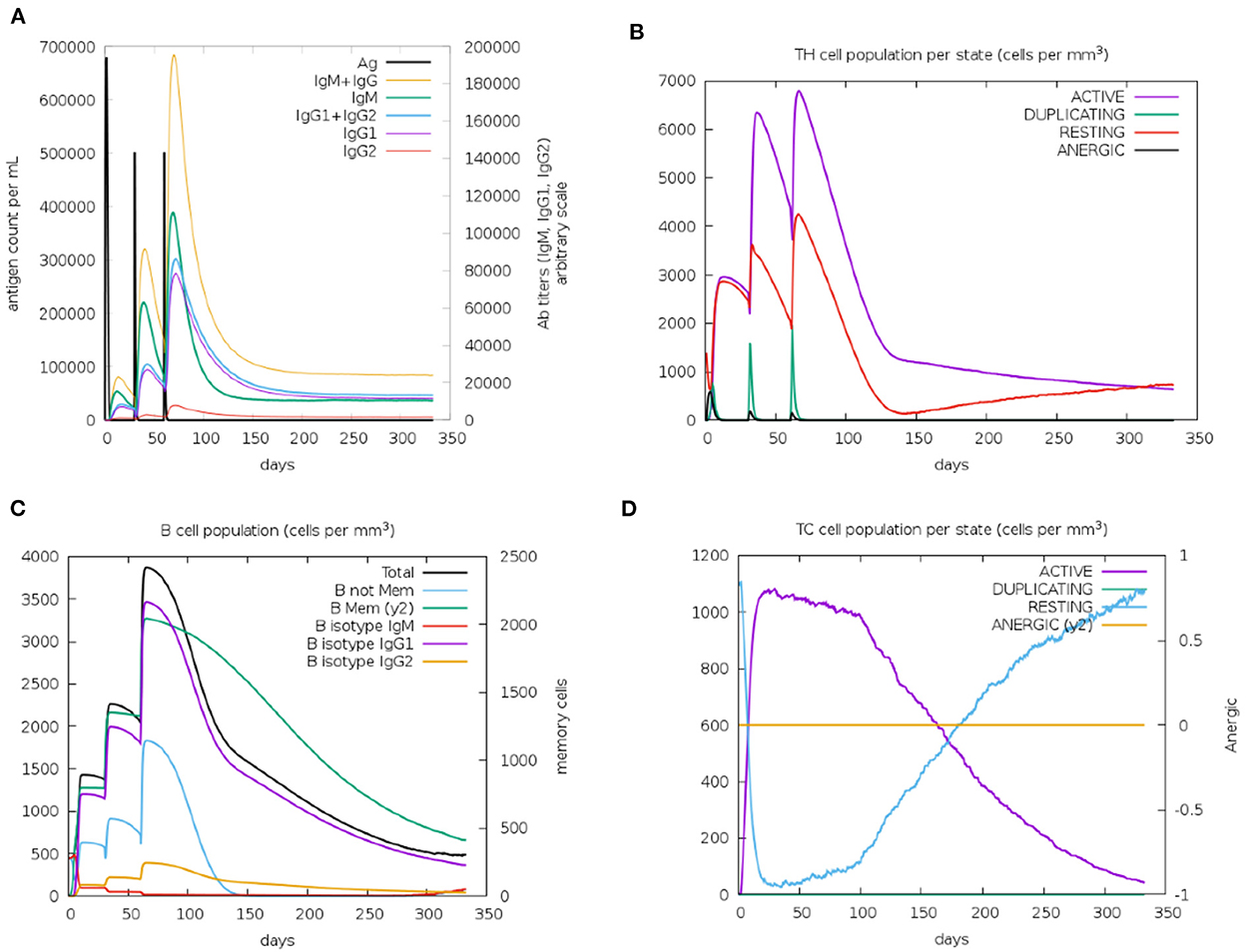 Figura 9. La inmunidad simulada de las construcciones de la vacuna utilizando el servidor C-ImmSim muestra (A) un aumento de las inmunoglobulinas después de la vacunación (B,D), que la población de linfocitos T citotóxicos y linfocitos T auxiliares aumenta después de la vacunación y se mantiene más alta durante todo el tiempo de exposición, y (C) un aumento de las poblaciones de células B después de la vacunación, produciendo altos niveles de inmunoglobulinas de memoria.
Figura 9. La inmunidad simulada de las construcciones de la vacuna utilizando el servidor C-ImmSim muestra (A) un aumento de las inmunoglobulinas después de la vacunación (B,D), que la población de linfocitos T citotóxicos y linfocitos T auxiliares aumenta después de la vacunación y se mantiene más alta durante todo el tiempo de exposición, y (C) un aumento de las poblaciones de células B después de la vacunación, produciendo altos niveles de inmunoglobulinas de memoria.
Optimización de codones y clonación in silico
Se utilizó la herramienta Java Codon Adaptation Tool para la optimización de codones. La secuencia de la proteína de la vacuna se tradujo inversamente para una expresión óptima en E. coli (cepa K12). La secuencia de ADN optimizada tuvo un valor de índice de adaptación de codones (CAI) de 1 y un contenido de GC de 54,05%, lo que indica que la vacuna diseñada se expresa teóricamente de manera estable en los huéspedes microbianos seleccionados. Además, la secuencia de ADN para la clonación en el vector de E. coli pET-28a(+) se diseñó utilizando el software SnapGene para la construcción de plásmidos recombinantes (Figura 10).
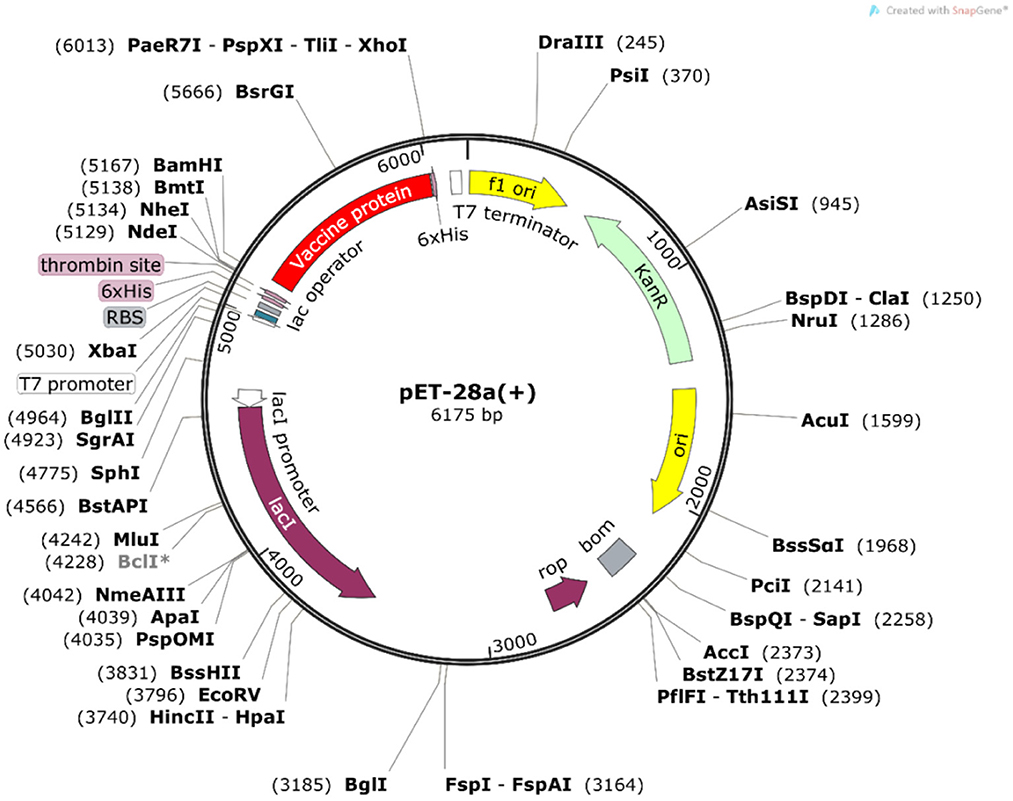 Figura 10. Clonación in silico de la construcción de la vacuna. La secuencia de ADN optimizada para codones de la construcción de la vacuna (en rojo) se clonó entre los sitios de restricción BamH I y Xho I del vector de expresión pET-28a(+) (que se muestra en círculos negros).
Figura 10. Clonación in silico de la construcción de la vacuna. La secuencia de ADN optimizada para codones de la construcción de la vacuna (en rojo) se clonó entre los sitios de restricción BamH I y Xho I del vector de expresión pET-28a(+) (que se muestra en círculos negros).
Expresión inducible y purificación de proteínas vacunales
Se añadió IPTG a E. coli BL21 (DE3) para inducir la expresión de pET-28a(+)-MEV, y el producto se examinó mediante electroforesis en gel SDS-PAGE. Los resultados mostraron que el recombinante pET-28a(+)-MEV inducido produjo una banda proteica específica alrededor de 33 kD como se esperaba (Figura 11), mientras que la muestra no inducida no mostró dicha banda. Los lisados sonicados se purificaron utilizando un Ni2+-Columna de resina NTA, el eluido se recolectó y concentró utilizando un filtro centrífugo Amicon Ultra-4 10K, y la proteína final de la vacuna se almacenó en glicerol al 10% a -80 °C.®
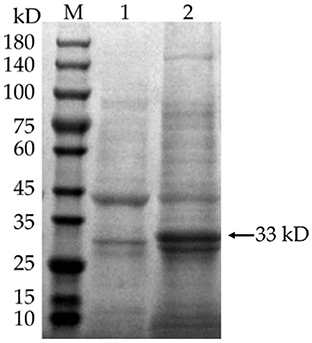 Figura 11. Expresión inducida de la proteína de la vacuna. M: estándar de calidad molecular de proteínas (10-180 kD); 1: bacterias recombinantes pET-28a(+)-MEV, no inducidas; 2: bacterias recombinantes pET-28a(+)-MEV, inducidas. Los geles de longitud completa se presentan en la Imagen Suplementaria 1.
Figura 11. Expresión inducida de la proteína de la vacuna. M: estándar de calidad molecular de proteínas (10-180 kD); 1: bacterias recombinantes pET-28a(+)-MEV, no inducidas; 2: bacterias recombinantes pET-28a(+)-MEV, inducidas. Los geles de longitud completa se presentan en la Imagen Suplementaria 1.
ELISA de 16 aislamientos clínicos HPS
Se utilizaron cultivos puros de cada uno de los 16 aislados clínicos de HPS para recubrir placas marcadas con enzimas a la misma concentración y volumen, y se realizaron ensayos ELISA utilizando anticuerpos policlonales de ratón obtenidos por inmunización con una vacuna multiepítopo, con un control sérico negativo. Los resultados se muestran en la Tabla 4 y en la Figura 12: los anticuerpos policlonales de ratón obtenidos fueron capaces de unirse a los 16 aislados clínicos de HPS, incluidos siete serotipos 5, dos serotipos 10, un serotipo 1, dos serotipos 7 y cuatro cepas no tipificables, y se realizó una prueba t de heterocedasticidad de una cola en los valores de OD450 de cada aislado frente a los correspondientes valores de OD450 séricos negativos, siendo todos los valores p menores que el nivel de prueba (α = 0,05), lo que indica que las diferencias fueron significativas. Estos resultados indican que los anticuerpos obtenidos de ratones inmunizados con la vacuna multiepítopo diseñada en este estudio fueron capaces de unirse a diferentes aislados de HPS serotipificados o no tipificables.
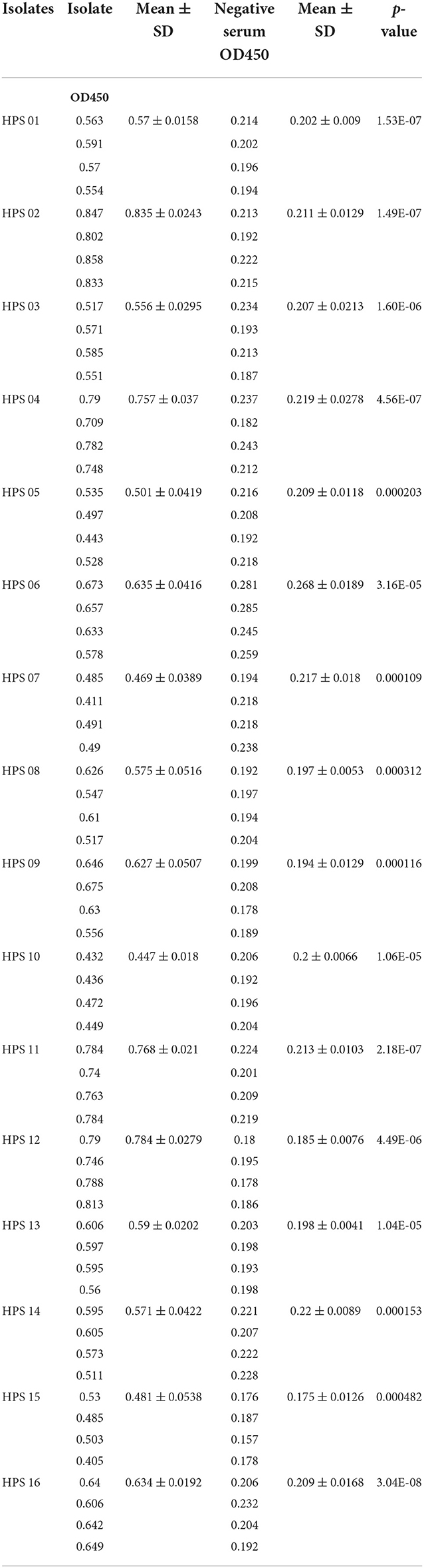 Tabla 4. Resultados de ELISA para cada aislado.
Tabla 4. Resultados de ELISA para cada aislado.
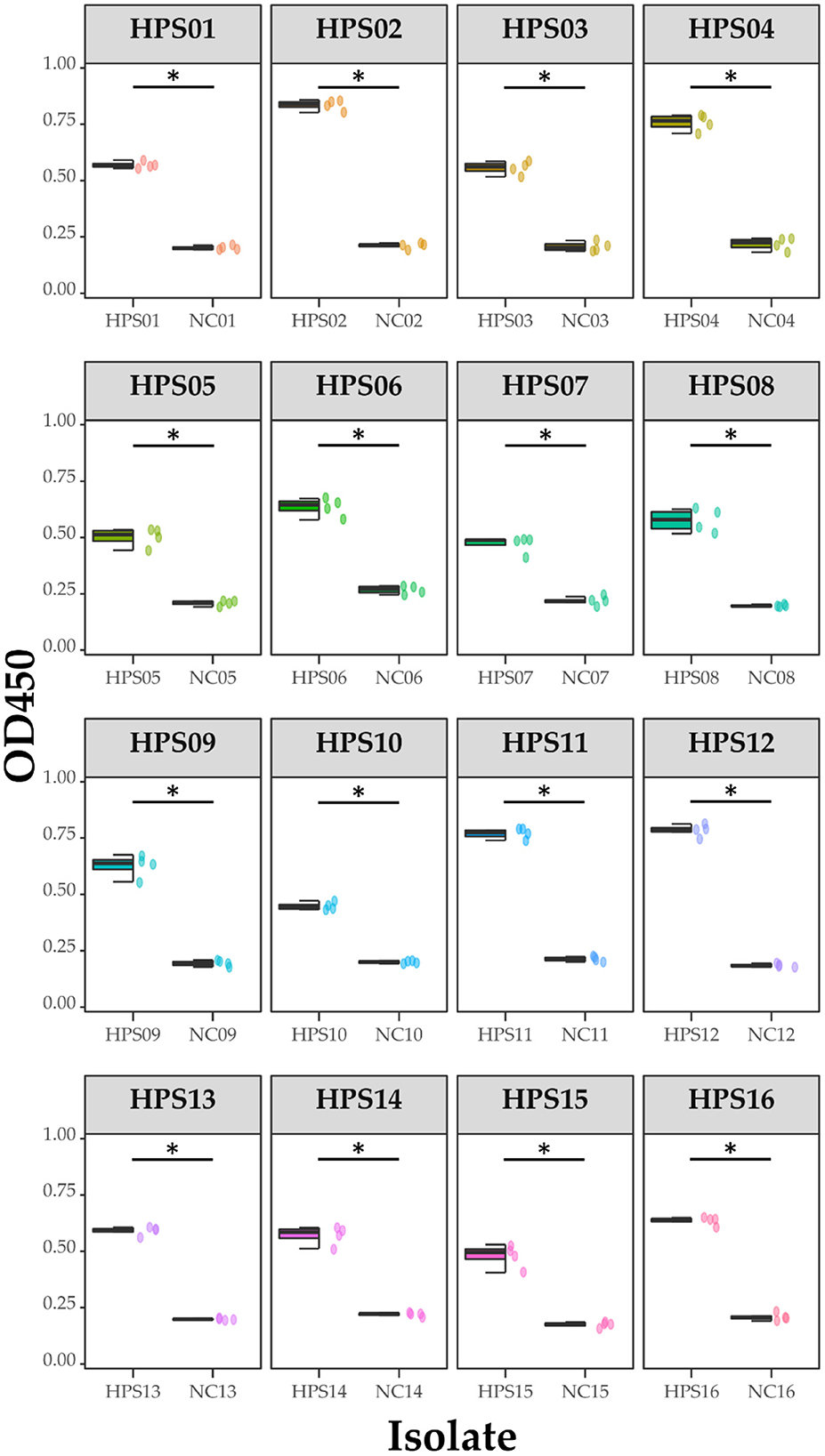 Figura 12. Resultados de ELISA para cada aislado. Los datos se presentan mediante diagramas de caja y dispersión; NC es un control sérico negativo; * indica diferencia estadísticamente significativa.
Figura 12. Resultados de ELISA para cada aislado. Los datos se presentan mediante diagramas de caja y dispersión; NC es un control sérico negativo; * indica diferencia estadísticamente significativa.
Discusión
Haemophilus parasuis causa principalmente la enfermedad de Glässer, caracterizada por poliserositis fibrinosa y artritis en cerdos de destete y que afecta a cerdos y cerdas en crecimiento. La vacunación es una medida eficaz para prevenir la mortalidad (36, 37). Sin embargo, las vacunas comerciales disponibles solo brindan protección contra un número limitado de serotipos. Aunque las vacunas autógenas son muy eficaces para proteger a los cerdos susceptibles, las cepas utilizadas para preparar las vacunas no se aíslan hasta después de un brote de la enfermedad (38). Además, para el tratamiento de la infección por HPS, el momento de la administración de antibióticos es crítico, y se debe administrar un tratamiento eficaz antes de que se desarrolle la inflamación fibrinosa. Sin embargo, debido a la dificultad del diagnóstico precoz, los cerdos enfermos suelen haber desarrollado una inflamación fibrinosa grave en el momento del diagnóstico. Incluso si el patógeno ha sido eliminado por los antibióticos, los cerdos pueden morir debido a la tormenta de citoquinas inducida por la inflamación y al daño histopatológico irreversible (39), y el tratamiento con antibióticos convencional suele ser ineficaz. Por lo tanto, este estudio tiene como objetivo diseñar una vacuna multiepítopo que se espera que proporcione una amplia protección contra la infección por HPS en granjas porcinas (todos los serotipos y cepas no tipificables). Se utilizó el análisis pangenómico para identificar el genoma central de HPS, se utilizaron programas de software y bases de datos para predecir los epítopos antigénicos de las proteínas de la membrana externa en el genoma central, y se utilizaron técnicas de vacunología inversa para diseñar la vacuna final de múltiples epítopos, reduciendo las prácticas experimentales tradicionales basadas en laboratorios.
En este trabajo, primero secuenciamos los genomas de 16 cepas de HPS utilizando la tecnología de secuenciación de nanoporos de Oxford (ONT) y obtuvimos ensamblajes completos de alta calidad. En comparación con las plataformas de secuenciación de segunda generación, la plataforma ONT tiene las ventajas de un menor coste, una velocidad de secuenciación más rápida, una mayor longitud de lectura y una mayor facilidad de funcionamiento (40). Posteriormente, los 16 genomas HPS obtenidos junto con los 105 genomas indexados por el NCBI se utilizaron para construir el pangenoma HPS utilizando el software Roary. Los resultados del análisis mostraron que HPS tiene un pangenoma abierto. El número de genes centrales representa solo el 14% del CDS de un genoma individual, y el resto es muy variable. La gran variación en los genomas de diferentes aislados de HPS es la razón principal de la falta de protección cruzada de las vacunas existentes. También vale la pena aclarar que no solo se utilizaron cepas virulentas, sino también cepas no virulentas para construir el pangenoma. La razón de esto es que las cepas no virulentas tienen el potencial de convertirse en cepas virulentas. Por ejemplo, se consideró que todas las cepas de serovar 7 no eran virulentas, pero algunas cepas de serovar 7 se han aislado de lesiones sistémicas de la enfermedad de Glässer, y la enfermedad se ha reproducido con una de ellas (41, 42).
Las siguientes ocho proteínas de la membrana externa se identificaron a partir de genes centrales y de núcleo blando mediante la predicción de péptidos señal y la localización subcelular: (1) Proteína de exportación de podoconjugados kpsD: esta proteína está involucrada principalmente en la translocación de podoconjugados desde el sitio de síntesis hasta la superficie celular (43). (2) Ensamblador de proteínas de membrana externa bamA: bamA está involucrado principalmente en catalizar el ensamblaje de proteínas transmembrana bacterianas y podría ser un candidato potencial a vacuna para la prevención de infecciones por Escherichia coli y Salmonella (44, 45). (3) Proteína de ensamblaje de lipopolisacáridos lptD: esta proteína media el transporte de lipopolisacáridos y el lptD de Vibrio Parahemolyticus es altamente inmunogénico, proporciona una protección del 100% contra la infección por Vibrio en ratones y es un antígeno potencial de la vacuna (46). (4) Lipoproteína Pal asociada a peptidoglicano: esta proteína interactúa con Tol Pal, es un agonista natural de TLR2 y se une fuertemente al LPS, que se libera en el torrente sanguíneo durante la infección que causa sepsis (47, 48). Además, se demostró que Pal de Legionella pneumophila, Haemophilus influenzae y Campylobacter jejuni es altamente inmunogénico y capaz de inducir respuestas inmunitarias innatas y adaptativas tempranas (49-51). (5) Lipoproteína mlaA: esta proteína, que pertenece a la misma clase de lipoproteínas que Pal, también está involucrada en el mantenimiento de la asimetría lipídica de la membrana externa de las bacterias Gram-negativas, formando una barrera osmótica para evitar la entrada de moléculas tóxicas (por ejemplo, antibióticos, desinfectantes, etc.). (6) La proteína del complejo de ensamblaje de autotransporte TamA: los estudios sugieren que esta proteína puede estar involucrada como sustrato para la secreción para facilitar la secreción de proteínas de autotransporte en lugar de en un sistema de autotransporte para la colonización de patógenos en el huésped (52). (7) Proteína activadora de la urea hidrolasa nlpD: esta proteína también es una lipoproteína, y en Cronobacter sakazakii, la nlpD responde al estrés ácido para resistir la fagocitosis manteniendo la integridad de la membrana. Además, la NLPD también puede estar involucrada en la regulación de la absorción de hierro y la actividad del sistema bis-arginina (53, 54). (8) Proteína de poro GBP: las proteínas de los poros están abundantemente presentes en la superficie de las bacterias como una barrera de tamizado y desempeñan un papel importante en las interacciones huésped-bacteria, lo que las convierte en posibles antígenos candidatos a vacunas y objetivos terapéuticos (55). A continuación, se identificaron epítopos de células B (n = 5), epítopos CTL (n = 3) y epítopos HTL (n = 6) a partir de estas proteínas de membrana externa para su uso en la construcción de la vacuna. Para mejorar la estabilidad de la estructura de la vacuna, los epítopos se unieron utilizando el enlazador EAAAK y el enlazador GPGPG (56, 57). A continuación, la vacuna multiepítopo se unió a la proteína modulina α4 soluble en fenol (un agonista de TLR 2) seleccionada como adyuvante utilizando un enlazador AYY para mejorar la inmunogenicidad de la vacuna (19). Posteriormente, se probó la antigenicidad y la alergenicidad de la construcción de la vacuna y se demostró que era antigénica y no alergénica con o sin relación con el adyuvante; Se predijo una puntuación de antigenicidad más alta para el acoplamiento con el adyuvante.
La construcción final de la vacuna que contiene epítopos de células B, epítopos CTL y epítopos HTL, así como enlazadores y adyuvante, tiene 280 residuos de aminoácidos y un peso molecular de 29,38 kD. El pI teórico de esta proteína vacunal fue de 9,65, lo que indica la naturaleza básica de la proteína. El índice de inestabilidad de la proteína es de 24,99, por lo que un valor de <40 indica que la construcción de la vacuna será estable siempre que se exprese (58). Además, otros indicadores demostraron la alta termoestabilidad, la naturaleza hidrofílica y la solubilidad de la construcción de la vacuna.
La predicción de la estructura secundaria de las construcciones de la vacuna mostró que solo el 2% de los residuos de aminoácidos estaban desordenados, lo que confirma la estabilidad de las construcciones. La construcción está compuesta principalmente por bobinas (73%), lo que facilitará la producción de anticuerpos. Además, se modeló y validó la construcción de la vacuna, y se investigó la interacción de la construcción de la vacuna con TLR2 mediante simulaciones de acoplamiento molecular para dilucidar una respuesta inmunitaria eficaz. La validación de la estructura 3D mostró que todos los residuos estaban en las regiones permitidas, y más del 90% estaban presentes en regiones favorables, lo que confirma que obtuvimos un modelo estructural de alta calidad. Además, el constructo tenía una alta afinidad de unión a TLR2, lo que indica que tiene el potencial de estimular la generación de una respuesta inmune. De acuerdo con los resultados de la simulación inmune del servidor C-ImmSim, se pudo observar un alto nivel de formación de células B de memoria y producción de anticuerpos, así como un mantenimiento mayor y prolongado de linfocitos T citotóxicos y auxiliares después de múltiples inmunizaciones, creando así una respuesta inmune humoral y celular que ayudará a prevenir infecciones. Además, la optimización de codones se realizó después de la traducción inversa de la proteína de la vacuna. Los valores de GC y CAI predichos para la proteína de la vacuna fueron 54,05% y 1 respectivamente, lo que indica que la proteína puede expresarse en grandes cantidades en E. coli.
La proteína de la vacuna se tradujo inversamente y se optimizó el codón, y se construyó el vector de expresión de E. coli pET-28a(+)-MEV y se transfirió a E. coli BL21 (DE3) para la inducción de la expresión, y se purificó la proteína diana de la vacuna. Los anticuerpos policlonales obtenidos de ratones inmunizados con la proteína de la vacuna fueron capaces de unirse a diferentes serotipos o aislados de HPS no tipificables, lo que indica preliminarmente que la proteína de la vacuna es una candidata vacunal prometedora. La vacuna multiepítopo diseñada en este estudio también tiene múltiples estrategias de uso clínico basadas en la infección y las características epidemiológicas del SPH, por ejemplo, en la inmunización directa de lechones contra la infección por SPH. Además, debido a que la cepa virulenta de HPS puede colonizar el tracto respiratorio de los lechones bajo la protección de anticuerpos maternos y así estimular al organismo del lechón para que produzca una respuesta inmune para prevenir la morbilidad y la mortalidad, este proceso se limita a la cepa a la que la cerda ha estado expuesta. Por lo tanto, una estrategia de inmunización alternativa que puede ser más eficaz es el uso de vacunas multiepítopo para inmunizar a las cerdas de reserva o gestantes para estimular la producción de anticuerpos contra varios serotipos o HPS no tipificables, con los lechones protegidos por anticuerpos maternos que exhiben una amplia gama de reactividades y colonizados por diferentes cepas de virulencia HPS. produciendo así una respuesta inmune para prevenir la infección.
Conclusión
El desarrollo de una nueva vacuna es necesario para abordar la compleja situación epidemiológica de Haemophilus parasuis y para resolver los problemas asociados a las vacunas existentes. En este estudio, utilizamos el análisis pangenómico con tecnología de vacuna inversa para construir una vacuna con el potencial de prevenir la infección por todos los serotipos, así como por Haemophilus parasuis no tipificable, y este proceso evita los inconvenientes de alto costo y tiempo del desarrollo de vacunas tradicionales. La construcción de la vacuna tenía múltiples epítopos de células B y T y exhibía alta antigenicidad, no toxicidad y no alergenicidad. Además, los resultados de la simulación inmunitaria mostraron que la vacuna activaba altos niveles de respuestas inmunitarias humorales y celulares. Los anticuerpos obtenidos de ratones inmunizados con la vacuna multiepítopo fueron capaces de unirse a diferentes aislados de HPS serotipificados o no tipificables. En conclusión, la vacuna diseñada en este estudio es una candidata prometedora para el control de Haemophilus parasuis.
Declaración de disponibilidad de datos
Las contribuciones originales presentadas en el estudio están incluidas en el artículo/Material complementario, las consultas posteriores pueden dirigirse al autor correspondiente.
Declaración ética
El estudio en animales fue revisado y aprobado por el Comité Provincial de Manejo de Animales de Laboratorio de Sichuan.
Contribuciones de los autores
MP y TT: metodología. MP y MR: software. PZ y TT: validación. PZ e YL: curación de datos. MP: redacción de la redacción del borrador original. TT y YW: redacción-revisión y edición. XY y ZY: administración de proyectos. Todos los autores han leído y aceptado la versión publicada del manuscrito.
Financiación
Este trabajo fue financiado por el Proyecto de Planificación de Ciencia y Tecnología de la Provincia de Sichuan (Número de proyecto: 2020YJ0345; Título: Mecanismo de Regulación Molecular del Sistema Antitoxina Toxina Tipo II de Haemophilus Parasuis en la Formación de Biopelículas Bacterianas); Proyecto de Planificación de Ciencia y Tecnología de la Provincia de Sichuan (Número de proyecto: 2021YFSY0005; Título: Desarrollo de una vacuna sinergista contra la enfermedad porcina basada en polisacáridos y citoquinas vegetales); Proyecto de Planificación de Ciencia y Tecnología de la Provincia de Sichuan (Número de proyecto: 2021YJ0270; Título: Establecimiento de un método de detección para la identificación de la infección por el virus de la peste porcina africana y su aplicación en el sistema de control de bioseguridad.
Reconocimientos
Agradecemos a YW por sus perspicaces comentarios sobre el diseño del estudio.
Conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un posible conflicto de intereses.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o afirmación que pueda ser hecha por su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fvets.2022.1053198/full#supplementary-material
Referencias
1. Brockmeier SL, Register KB, Kuehn JS, Nicholson TL, Loving CL, Bayles DO, et al. Virulencia y borrador de la secuencia del genoma de múltiples cepas del patógeno porcino Haemophilus parasuis. PLoS UNO. (2014) 9:E103787. doi: 10.1371/journal.pone.0103787
Resumen de PubMed | Texto completo de CrossRef | Google Académico
2. Kielstein P, Rapp-Gabrielson VJ. Designación de 15 serovares de Haemophilus parasuis sobre la base de la inmunodifusión mediante extractos de antígenos termoestables. J Clin Microbiol. (1992) 30:862–5. doi: 10.1128/jcm.30.4.862-865.1992
Resumen de PubMed | Texto completo de CrossRef | Google Académico
3. Blackall PJ, Trott DJ, Rapp-Gabrielson V, Hampson DJ. Análisis de Haemophilus parasuis mediante electroforesis enzimática multilocus. Microbiol veterinario. (1997) 56:125–34. doi: 10.1016/S0378-1135(96)01342-9
Resumen de PubMed | Texto completo de CrossRef | Google Académico
4. Cerdà-Cuéllar M, Naranjo JF, Verge A, Nofrarías M, Cortey M, Olvera A, et al. La vacunación de cerdas modula la colonización de lechones por Haemophilus parasuis. Microbiol veterinario. (2010) 145:315–20. doi: 10.1016/j.vetmic.2010.04.002
Resumen de PubMed | Texto completo de CrossRef | Google Académico
5. Olvera A, Cerdà-Cuéllar M, Aragón V. Estudio de la estructura poblacional de Haemophilus parasuis mediante tipificación de secuencias multilocus. Microbiología. (2006) 152:3683–90. doi: 10.1099/mic.0.29254-0
Resumen de PubMed | Texto completo de CrossRef | Google Académico
6. Olvera A, Cerdà-Cuéllar M, Nofrarías M, Revilla E, Segalés J, Aragón V. Dinámica de genotipos de Haemophilus parasuis en una granja recuperada de un brote de la enfermedad de Glässer. Microbiol veterinario. (2007) 123:230–7. doi: 10.1016/j.vetmic.2007.03.004
Resumen de PubMed | Texto completo de CrossRef | Google Académico
7. Jalal K, Khan K, Ahmad D, Hayat A, Basharat Z, Abbas MN, et al. Enfoque de vacunología inversa pangenómica para el diseño de una vacuna multiepítopo contra Escherichia albertii. Int J Mol Sci. (2021) 22:12814. doi: 10.3390/ijms222312814
Resumen de PubMed | Texto completo de CrossRef | Google Académico
8. D’Mello A, Ahearn CP, Murphy TF, Tettelin H. ReVac: una línea computacional de vacunología inversa para la priorización de candidatos a vacunas de proteínas procariotas. BMC Genómica. (2019) 20:981. doi: 10.1186/s12864-019-6195-y
Resumen de PubMed | Texto completo de CrossRef | Google Académico
9. Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: ensamblaje escalable y preciso de lectura larga a través de la ponderación adaptativa k-mer y la separación de repeticiones. Genoma Res. (2017) 27:722–36. doi: 10.1101/gr.215087.116
Resumen de PubMed | Texto completo de CrossRef | Google Académico
10. Huang Y, Liu P, Shih P. Homopolish: un método para la eliminación de errores sistemáticos en la secuenciación de nanoporos mediante pulido homólogo. Genoma Biol. (2021) 22:95. doi: 10.1186/s13059-021-02282-6
Resumen de PubMed | Texto completo de CrossRef | Google Académico
11. Seemann T. Prokka: anotación rápida del genoma procariota. Bioinformática. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
Resumen de PubMed | Texto completo de CrossRef | Google Académico
12. Página AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: análisis rápido del genoma de la panola procariota a gran escala. Bioinformática. (2015) 31:3691–3. doi: 10.1093/bioinformatics/btv421
Resumen de PubMed | Texto completo de CrossRef | Google Académico
13. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: análisis de genética evolutiva molecular a través de plataformas informáticas. Mol Biol Evol. (2018) 35:1547–9. DOI: 10.1093/molbev/msy096
Resumen de PubMed | Texto completo de CrossRef | Google Académico
14. Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, et al. SignalP 50 mejora las predicciones de péptidos de señal mediante redes neuronales profundas. Nat Biotechnol. (2019) 37:420–3. doi: 10.1038/s41587-019-0036-z
Resumen de PubMed | Texto completo de CrossRef | Google Académico
15. Ong E, Wang H, Wong MU, Seetharaman M, Valdez N, He Y. Vaxign-ML: modelo de vacunología inversa de aprendizaje automático supervisado para mejorar la predicción de antígenos protectores bacterianos. Bioinformática. (2020) 36:3185–91. doi: 10.1093/bioinformatics/btaa119
Resumen de PubMed | Texto completo de CrossRef | Google Académico
16. Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: mejora de la predicción de epítopos de células B basada en secuencias mediante epítopos conformacionales. Ácidos nucleicos Res. (2017) 45:W24–9. doi: 10.1093/nar/gkx346
Resumen de PubMed | Texto completo de CrossRef | Google Académico
17. Peters B, Bulik S, Tampe R, Van Endert PM, Holzhütter H. Identificación de epítopos MHC de clase I mediante la predicción de la eficiencia de transporte TAP de los precursores de epítopos. J Immunol. (2003) 171:1741–9. doi: 10.4049/jimmunol.171.4.1741
Resumen de PubMed | Texto completo de CrossRef | Google Académico
18. Nielsen M, Lund O. NN-align: un algoritmo de alineación basado en redes neuronales artificiales para la predicción de la unión a péptidos MHC de clase II. BMC Bioinformat. (2009) 10:296. doi: 10.1186/1471-2105-10-296
Resumen de PubMed | Texto completo de CrossRef | Google Académico
19. Chatterjee R, Sahoo P, Mahapatra SR, Dey J, Ghosh M, Kushwaha GS, et al. Desarrollo de una vacuna quimérica conservada para la inducción de una fuerte respuesta inmune contra Staphylococcus aureus utilizando enfoques inmunoinformáticos. Vacunas. (2021) 9:1038. doi: 10.3390/vacunas9091038
Resumen de PubMed | Texto completo de CrossRef | Google Académico
20. Doytchinova IA, Flower DR. VaxiJen: un servidor para la predicción de antígenos protectores, antígenos tumorales y vacunas de subunidades. BMC Bioinformat. (2007) 8:4. doi: 10.1186/1471-2105-8-4
Resumen de PubMed | Texto completo de CrossRef | Google Académico
21. Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP v.2: un servidor para la predicción in silico de alérgenos. Modelo J Mol. (2014) 20:2278. doi: 10.1007/s00894-014-2278-5
Resumen de PubMed | Texto completo de CrossRef | Google Académico
22. Magnan CN, Randall A, Baldi P. SOLpro: predicción precisa basada en secuencias de la solubilidad de proteínas. Bioinformática. (2009) 25:2200–7. doi: 10.1093/bioinformatics/btp386
Resumen de PubMed | Texto completo de CrossRef | Google Académico
23. McGuffin LJ, Bryson K, Jones DT. El servidor de predicción de la estructura de la proteína PSIPRED. Bioinformática. (2000) 16:404–5. doi: 10.1093/bioinformatics/16.4.404
Resumen de PubMed | Texto completo de CrossRef | Google Académico
24. Wang S, Li W, Liu S, Xu J. RaptorX-Property: un servidor web para la predicción de propiedades de la estructura de proteínas. Ácidos nucleicos Res. (2016) 44:W430–5. doi: 10.1093/nar/gkw306
Resumen de PubMed | Texto completo de CrossRef | Google Académico
25. Kim DE, Chivian D, Baker D. Predicción y análisis de la estructura de proteínas utilizando el servidor Robetta. Ácidos nucleicos Res. (2004) 32:W526-31. doi: 10.1093/nar/gkh468
Resumen de PubMed | Texto completo de CrossRef | Google Académico
26. Colovos C, Yeates TO. Verificación de estructuras proteicas: patrones de interacciones atómicas no enlazadas. Ciencia de proteínas. (1993) 2:1511–9. doi: 10.1002/pro.5560020916
Resumen de PubMed | Texto completo de CrossRef | Google Académico
27. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: un programa para comprobar la calidad estereoquímica de las estructuras de las proteínas. J Appl Crystallogr. (1993) 26:283–91. doi: 10.1107/S0021889892009944
28. Ponomarenko J, Bui H, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: una nueva herramienta basada en la estructura para la predicción de epítopos de anticuerpos. BMC Bioinformat. (2008) 9:514. doi: 10.1186/1471-2105-9-514
Resumen de PubMed | Texto completo de CrossRef | Google Académico
29. Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. El servidor web ClusPro para el acoplamiento proteína-proteína. Nat Protoc. (2017) 12:255–78. doi: 10.1038/nprot.2016.169
Resumen de PubMed | Texto completo de CrossRef | Google Académico
30. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. El banco de datos de proteínas. Ácidos nucleicos Res. (2000) 28:235–42. doi: 10.1093/nar/28.1.235
Resumen de PubMed | Texto completo de CrossRef | Google Académico
31. Rapin N, Lund O, Bernaschi M, Castiglione F. La inmunología computacional se encuentra con la bioinformática: el uso de herramientas de predicción para la unión molecular en la simulación del sistema inmunológico. PLoS UNO. (2010) 5:e9862. doi: 10.1371/journal.pone.0009862
Resumen de PubMed | Texto completo de CrossRef | Google Académico
32. Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, et al. JCat: una herramienta novedosa para adaptar el uso de codones de un gen diana a su huésped de expresión potencial. Ácidos nucleicos Res. (2005) 33:W526–31. doi: 10.1093/nar/gki376
Resumen de PubMed | Texto completo de CrossRef | Google Académico
33. Chen FZ, You LJ, Yang F, Wang LN, Guo XQ, Gao F, et al. CNGBdb: Base de datos del banco nacional de germoplasma de China. Yi chuan Hereditas. (2020) 42:799–809.
34. Guo X, Chen F, Gao F, Li L, Liu K, You L, et al. CNSA: un repositorio de datos para archivar datos ómicos. Base de datos. (2020) 2020:baaa055. doi: 10.1093/database/baaa055
Resumen de PubMed | Texto completo de CrossRef | Google Académico
35. Majid M, Andleeb S. Diseño de una vacuna multiepitópica contra el enterotoxigénico Bacteroides fragilis basada en un enfoque inmunoinformático. Sci Rep. (2019) 9:19780. doi: 10.1038/s41598-019-55613-w
Resumen de PubMed | Texto completo de CrossRef | Google Académico
36. Martín de la Fuente AJ, Rodríguez-Ferri EF, Frandoloso R, Martínez S, Tejerina F, Gutiérrez-Martín CB. Respuesta sistémica de anticuerpos en cerdos privados de calostro infectados experimentalmente con Haemophilus parasuis. Res Vet Sci. (2009) 86:248–53. doi: 10.1016/j.rvsc.2008.07.017
Resumen de PubMed | Texto completo de CrossRef | Google Académico
37. Takahashi K, Naga S, Yagihashi T, Ikehata T, Nakano Y, Senna K, et al. Un experimento de protección cruzada en cerdos vacunados con bacterinas 2 y 5 de los serovares Haemophilus parasuis, y evaluación de una vacuna bivalente en condiciones de laboratorio y de campo. J Vet Medical Sci. (2001) 63:487–91. doi: 10.1292/jvms.63.487
Resumen de PubMed | Texto completo de CrossRef | Google Académico
38. McOrist S, Bowles R, Blackall P. Vacunación autógena de cerdas para la enfermedad de Glasser en cerdos destetados en dos grandes sistemas de granjas porcinas. J Salud Porcina Prod. (2009) 17:90–6.
39. Costa-Hurtado M, Olvera A, Martinez-Moliner V, Galofré-Milà N, Martínez P, Domínguez J, et al. Cambios en el fenotipo de macrófagos después de la infección de cerdos con cepas de Haemophilus parasuis con diferentes niveles de virulencia. Infectar a Immun. (2013) 81:2327–33. doi: 10.1128/IAI.00056-13
Resumen de PubMed | Texto completo de CrossRef | Google Académico
40. Zhang P, Jiang D, Wang Y, Yao X, Luo Y, Yang Z. Comparación de estrategias de ensamblaje de novo para genomas bacterianos. Int J Mol Sci. (2021) 22:14. doi: 10.3390/ijms22147668
Resumen de PubMed | Texto completo de CrossRef | Google Académico
41. Aragón V, Cerdà-Cuéllar M, Fraile L, Mombarg M, Nofrarías M, Olvera A, et al. Correlación entre el resultado clínico-patológico y la tipificación de las cepas de campo de Haemophilus parasuis. Microbiol veterinario. (2010) 142:387–93. doi: 10.1016/j.vetmic.2009.10.025
Resumen de PubMed | Texto completo de CrossRef | Google Académico
42. Dazzi CC, Guizzo JA, Prigol SR, Kreutz LC, Driemeier D, Chaudhuri S, et al. Nuevas lesiones patológicas desarrolladas en cerdos por una cepa «no virulenta» de Glaesserella parasuis. Front Vet Sci. (2020) 7:98. doi: 10.3389/fvets.2020.00098
Resumen de PubMed | Texto completo de CrossRef | Google Académico
43. Arrecubieta C, Hammarton TC, Barrett B, Chareonsudjai S, Hodson N, Rainey D, et al. El transporte de polisacáridos capsulares del grupo 2 a través del espacio periplásmico en Escherichia coli. Funciones de las proteínas KpsE y KpsD. J Biol Chem. (2001) 276:4245–50. doi: 10.1074/jbc. M008183200
Resumen de PubMed | Texto completo de CrossRef | Google Académico
44. Guan Q, Wang X, Wang X, Teng D, Wang J. Análisis in silico y expresión recombinante de la proteína BamA como vacuna universal contra Escherichia coli en ratones. Aplicación Microbiol Biot. (2016) 100:5089–98. doi: 10.1007/s00253-016-7467-y
Resumen de PubMed | Texto completo de CrossRef | Google Académico
45. Konovalova A, Kahne DE, Silhavy TJ. Biogénesis de la membrana externa. Annu Rev Microbiol. (2017) 71:539–56. doi: 10.1146/annurev-micro-090816-093754
Resumen de PubMed | Texto completo de CrossRef | Google Académico
46. Zha Z, Li C, Li W, Ye Z, Pan J. LptD es un antígeno vacunal prometedor y una posible diana inmunoterapéutica para la protección contra la infección por especies de Vibrio. Sci Rep. (2016) 6:38577. doi: 10.1038/srep38577
Resumen de PubMed | Texto completo de CrossRef | Google Académico
47. Godlewska R, Wiśniewska K, Pietras Z, Jagusztyn-Krynicka EK. Lipoproteína asociada a peptidoglicano (Pal) de bacterias gramnegativas: función, estructura, papel en la patogénesis y posible aplicación en inmunoprofilaxis. FEMS Microbiol Lett. (2009) 298:1–11. doi: 10.1111/j.1574-6968.2009.01659.x
Resumen de PubMed | Texto completo de CrossRef | Google Académico
48. Liang MD, Bagchi A, Warren HS, Tehan MM, Trigilio JA, Beasley-Topliffe LK, et al. Lipoproteína bacteriana asociada al peptidoglicano: un agonista natural del receptor tipo Toll 2 que se libera en el suero y tiene sinergia con el lipopolisacárido. J Infectar Dis. (2005) 191:939–48. doi: 10.1086/427815
Resumen de PubMed | Texto completo de CrossRef | Google Académico
49. Mobarez AM, Rajabi RA, Salmanian AH, Khoramabadi N, Hosseini Doust SR. Inducción de inmunidad protectora por la proteína lipoproteína asociada a peptidoglicano recombinante (rPAL) de Legionella pneumophila en un modelo de ratón BALB/c. Patogénesis microb. (2019) 128:100–5. doi: 10.1016/j.micpath.2018.12.014
Resumen de PubMed | Texto completo de CrossRef | Google Académico
50. Kodama S, Hirano T, Suenaga S, Abe N, Suzuki M. La trompa de Eustaquio posee características inmunológicas como sitio efector de la mucosa y responde a la proteína de membrana externa P6 de Haemophilus influenzae no tipificable. Vacuna. (2006) 24:1016–27. doi: 10.1016/j.vaccine.2005.07.110
Resumen de PubMed | Texto completo de CrossRef | Google Académico
51. Cordwell SJ, Len ACL, Touma RG, Scott NE, Falconer L, Jones D, et al. Identificación de proteínas asociadas a membranas de cepas de Campylobacter jejuni utilizando tecnologías proteómicas complementarias. Proteómica. (2008) 8:122–39. doi: 10.1002/pmic.200700561
Resumen de PubMed | Texto completo de CrossRef | Google Académico
52. Selkrig J, Mosbahi K, Webb CT, Belousoff MJ, Perry AJ, Wells TJ, et al. Descubrimiento de un sistema arquetípico de transporte de proteínas en las membranas externas bacterianas. Nat Struct Mol Biol. (2012) 19:506–10. doi: 10.1038/nsmb.2261
Resumen de PubMed | Texto completo de CrossRef | Google Académico
53. Ji X, Lu P, Xue J, Zhao N, Zhang Y, Dong L, et al. La lipoproteína NlpD en Cronobacter sakazakii responde al estrés ácido y regula la resistencia y virulencia de los macrófagos manteniendo la integridad de la membrana. Virulencia. (2021) 12:415–29. doi: 10.1080/21505594.2020.1870336
Resumen de PubMed | Texto completo de CrossRef | Google Académico
54. Tidhar A, Levy Y, Zauberman A, Vagima Y, Gur D, Aftalion M, et al. La alteración de la lipoproteína NlpD del patógeno de la peste Yersinia pestis afecta a la adquisición de hierro y a la actividad del sistema de translocación de arginina gemela. Plos Negligencia Trop D. (2019) 13:E0007449. doi: 10.1371/journal.pntd.0007449
Resumen de PubMed | Texto completo de CrossRef | Google Académico
55. Sharma A, Yadav SP, Sarma D, Mukhopadhaya A. Modulación de las respuestas celulares del huésped por porinas bacterianas gramnegativas. Adv Protein Chem STR. (2022) 128:35–77. doi: 10.1016/bs.apcsb.2021.09.004
Resumen de PubMed | Texto completo de CrossRef | Google Académico
56. Chen X, Zaro JL, Shen W. Enlazadores de proteínas de fusión: propiedad, diseño y funcionalidad. (2013) 65:1357–69. doi: 10.1016/j.addr.2012.09.039
Resumen de PubMed | Texto completo de CrossRef | Google Académico
57. Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T. Diseño de los enlazadores que separan eficazmente los dominios de una proteína de fusión bifuncional. Ing. Proteica. (2001) 14:529–32. doi: 10.1093/proteína/14.8.529
Resumen de PubMed | Texto completo de CrossRef | Google Académico
58. Guruprasad K, Reddy BV, Pandit MW. Correlación entre la estabilidad de una proteína y su composición dipeptídica: un enfoque novedoso para predecir la estabilidad in vivo de una proteína a partir de su secuencia primaria. Ing. Proteica. (1990) 4:155–61. doi: 10.1093/proteína/4.2.155
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Palabras clave: Haemophilus parasuis, vacunología inversa, análisis pangenómico, vacuna multiepítopo, inmunoinformática
Cita: Pang M, Tu T, Wang Y, Zhang P, Ren M, Yao X, Luo Y y Yang Z (2022) Diseño de una vacuna multiepítopo contra Haemophilus parasuis basada en enfoques pangenómicos e inmunoinformáticos. Frente. Vet. Sci. 9:1053198. doi: 10.3389/fvets.2022.1053198
Recibido: 25 de septiembre de 2022; Aceptado: 30 de noviembre de 2022;
Publicado: 29 diciembre 2022.
Editado por:
Lian-Feng Li, Instituto de Investigación Veterinaria de Harbin (CAAS), China
Revisado por:
Yuexiu Zhang, Universidad Estatal de Ohio, Estados
Unidos Francesco Ria, Universidad Católica del Sagrado Corazón, Italia
Derechos de autor © 2022 Pang, Tu, Wang, Zhang, Ren, Yao, Luo y Yang. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia Creative Commons Attribution License (CC BY). S
*Correspondencia: Yin Wang, 10334@sicau.edu.cn
†Estos autores han contribuido igualmente a este trabajo y comparten la primera autoría
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, ni las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o afirmación que pueda hacer su fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía