
La construcción de una red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA identifica genes y vías relacionadas con la fertilidad de las cabras

La construcción de una red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA identifica genes y vías relacionadas con la fertilidad de las cabras
 Farzad Ghafouri1†,
Farzad Ghafouri1†,  Mostafa Sadeghi1*†,
Mostafa Sadeghi1*†,  Abolfazl Bahrami1,2*†, Masoumeh Naserkheil1,3, Vahid Dehghanian Reyhan1, Arash Javanmard4, Seyed Reza Miraei-Ashtiani1, Soheila Ghahremani5,
Abolfazl Bahrami1,2*†, Masoumeh Naserkheil1,3, Vahid Dehghanian Reyhan1, Arash Javanmard4, Seyed Reza Miraei-Ashtiani1, Soheila Ghahremani5,  Herman W. Barkema6, Rostam Abdollahi-Arpanahi1 y
Herman W. Barkema6, Rostam Abdollahi-Arpanahi1 y  John P. Kastelic6
John P. Kastelic6- 1Departamento de Ciencia Animal, Facultad de Agricultura y Recursos Naturales, Universidad de Teherán, Karaj, Irán
- 2Centro Biomédico de Ciencias de Biología de Sistemas Munich, Universidad Ludwig-Maximilians, Munich, Alemania
- 3División de Cría y Genética Animal, Instituto Nacional de Ciencia Animal, Cheonan-si, República de Corea
- 4Departamento de Ciencias Animales, Facultad de Agricultura, Universidad de Tabriz, Tabriz, Irán
- 5Departamento de Ciencia Animal, Facultad de Agricultura, Universidad de Tarbiat Modares, Teherán, Irán
- 6Facultad de Medicina Veterinaria, Universidad de Calgary, Calgary, AB, Canadá
Fondo: Existe un creciente interés en la mejora genética de los rasgos de fertilidad en cabras hembras. Con el genotipado de alto rendimiento, la secuenciación de ARN unicelular (scRNA-seq) es una herramienta poderosa para medir perfiles de expresión génica. El objetivo principal fue investigar el perfil comparativo del transcriptoma de las células de la granulosa (GC) de cabras de alta y baja fertilidad, utilizando scRNA-seq.
Métodos: Treinta muestras de cabras Ji’ning Gray (n = 15 para alta fertilidad y n = 15 para baja fertilidad ) fueron recuperadas de datos de scRNA-seq disponibles públicamente. Se aplicó un análisis de enriquecimiento funcional y un enfoque de minería de literatura para explorar módulos y genes centrales relacionados con la fertilidad. Luego, se predijeron las interacciones entre los tipos de ARN identificados, y la red reguladora de ceRNA se construyó integrando estas interacciones con otras redes reguladoras de genes (GRN).
Resultados y discusión: Los análisis comparativos relacionados con la transcriptómica identificaron 150 genes expresados diferencialmente (DEG) entre grupos de fertilidad alta y baja, según el cambio de pliegue (≥5 y ≤−5) y la tasa de descubrimiento falso (FDR <0.05). Entre estos genes, 80 fueron regulados al alza y 70 fueron regulados a la baja. Además, se identificaron 81 ARNm, 58 circRNAs, 8 lincRNAs, 19 lncRNAs y 55 miRNAs mediante minería bibliográfica. Además, identificamos 18 genes hub (SMAD1, SMAD2, SMAD3, SMAD4, TIMP1, ERBB2, BMP15, TGFB1, MAPK3, CTNNB1, BMPR2, AMHR2, TGFBR2, BMP4, ESR1, BMPR1B, AR y TGFB2) implicado en la fertilidad caprina. Las redes y módulos biológicos identificados se asociaron principalmente con las vías de firma del ovario. Además, el análisis de enriquecimiento de KEGG identificó la regulación de la pluripotencia de las células madre, las interacciones citocina-receptor de citoquinas, la esteroidogénesis ovárica, la meiosis de ovocitos, la maduración de ovocitos mediada por progesterona, la síntesis paratiroidea y de la hormona del crecimiento, la síntesis y secreción de cortisol, y las vías de señalización para prolactina, TGF-beta, hipopótamo, MAPK, PI3K-Akt y FoxO. La anotación funcional de los DEG identificados implicó importantes vías biológicas. Estos hallazgos proporcionaron información sobre la base genética de la fertilidad en cabras hembras y son un impulso para dilucidar las redes reguladoras moleculares del ceRNA y las funciones de los DEG subyacentes al desarrollo folicular ovárico.
1 Introducción
La mejora en el rendimiento reproductivo es una prioridad en la industria caprina, ya que es uno de los determinantes más importantes de la productividad, la sostenibilidad y la rentabilidad. El ciclo reproductivo implica funciones ováricas dinámicas y complejas, caracterizadas por la aparición progresiva y el desarrollo de folículos ováricos (Fatet et al., 2011) bajo control endocrino. Tanto las células de la granulosa como las de la teca están involucradas en la esteroidogénesis (Qiu et al., 2013). Las células de la granulosa (GC) son uno de los tipos de células más importantes en los folículos ováricos, desempeñando un papel crucial en el desarrollo folicular y la atresia (Matsuda et al., 2012), especialmente en las últimas etapas del desarrollo de los ovocitos y la ovulación. Además, estas células también controlan la maduración citoplasmática y juegan un papel clave en la maduración nuclear al responder a las gonadotropinas (Mori et al., 2000). Por lo tanto, la función reproductiva en la hembra es inherentemente compleja, involucrando varios procesos anatómicos y fisiológicos (Li et al., 2021a). Además, el tamaño de la camada, un atributo importante directamente relacionado con la eficiencia reproductiva, está controlado por múltiples genes y factores (Lai et al., 2016). Por lo tanto, el conocimiento de la base genética de la eficiencia reproductiva proporcionará información sobre los componentes que controlan la foliculogénesis ovárica y la fertilidad en cabras (de Lima et al., 2020).
El enfoque genético candidato para la fertilidad se ha estudiado ampliamente en varias especies de ganado (Miao et al., 2016a; Miao et al., 2016b; Bahrami et al., 2017; Quan et al., 2019). Además, requiere herramientas bien desarrolladas para detectar y caracterizar múltiples genes, vías y redes (Ahlawat et al., 2016; Zhang et al., 2017; Ghafouri y otros, 2021; Naserkheil et al., 2022).
La secuenciación de ARN unicelular (scRNA-seq) se ha utilizado para caracterizar transcripciones y diferencias en la expresión génica, identificar genes funcionales y analizar redes reguladoras en numerosas especies (Chen et al., 2015; Li y otros, 2021b; Li et al., 2021c). Además, scRNA-seq se está utilizando para mapear y cuantificar la actividad transcripcional a resolución de una sola célula para todos los genes en el genoma (Islam et al., 2014). Es útil para el análisis de la heterogeneidad celular, ya que puede secuenciar simultáneamente miles de células y descubrir nuevos tipos de células en animales (Choi y Kim, 2019). Por el contrario, se necesita un enfoque integrado para gestionar los datos a gran escala generados con tecnologías de alto rendimiento junto con la minería de literatura. Los análisis integrados pueden combinar vistas multinivel de datos fisiológicos en una interpretación total de procedimientos moleculares reguladores no lineales (La et al., 2019; Reyhan y otros, 2022; Sadeghi et al., 2022). Actualmente, varias herramientas bioinformáticas, enfoques computacionales y algoritmos están disponibles para identificar interacciones y funciones de proteínas en módulos reguladores en varias redes biológicas complejas (Bugrim et al., 2004). En este sentido, las redes multipartitas como circRNA-lincRNA-lncRNA-miRNA-mRNA ceRNA redes reguladoras consideran varios ARN y han puesto de relieve un nuevo mecanismo regulador de interacción entre ARN. Los ARN circulares (circRNAs) son moléculas de ARN monocatenarias, cerradas covalentemente sin extremos libres de 5′ y 3′ que ejercen una función biológica al actuar como reguladores transcripcionales, esponjas de microARN y plantillas de proteínas (Zhou et al., 2020). Los ARN no codificantes intergénicos largos (lincRNAs), las transcripciones de ARN con >200 nucleótidos, desempeñan un papel importante en procesos biológicos como el control de la expresión génica, el control epigenético y la formación de andamios (Deniz y Erman, 2017). Los ARN largos no codificantes (lncRNA) son transcripciones de ARN no codificantes involucradas en diversos procedimientos biológicos, como la proliferación celular y la regulación transcripcional (Wei et al., 2016). Además, los microARN (miARN), moléculas reguladoras con 19-25 nucleótidos, desempeñan funciones reguladoras vitales en múltiples procedimientos biológicos (diferenciación y migración celular, oncogénesis y apoptosis) al suprimir los ARNm (Wang et al., 2015). Por lo tanto, este enfoque parece adecuado para comprender los mecanismos de regulación molecular en rasgos poligénicos (Hallock y Thomas, 2012).
Varios estudios han identificado importantes genes candidatos asociados con la regulación hormonal del ciclo reproductivo y los rasgos de fertilidad en cabras (Li et al., 2010; Ahlawat et al., 2016; Lai et al., 2016; Zhang et al., 2018a; Li et al., 2021b). Además, la ontología génica y la biología de sistemas permiten la identificación de genes centrales y genes de coexpresión con roles críticos en la fertilidad (Ahlawat et al., 2016; Zhang et al., 2018b). Por ejemplo, en un estudio que comparó cabras de alta fertilidad versus baja, se identificaron muchos genes candidatos en cada grupo. En un análisis del genoma completo de cabras lecheras chinas de Laoshan, varios genes candidatos (CCNB2, AR, SMAD2, AMHR2, KDM6A, SOX5 y SYCP2) se asociaron con una fertilidad alta y baja (Lai et al., 2016). Por lo tanto, el propósito del presente estudio fue examinar las regiones genómicas de la firma GC ovárica de cabras Ji’ning Gray con alta y baja fertilidad, utilizando un conjunto de datos scRNA-seq de repositorio público. Además, revisamos críticamente la literatura, buscando publicaciones relevantes utilizando palabras clave relacionadas con GC y fertilidad en cabras. Se compilaron genes con diferencias significativas relacionadas con nuestros análisis bioinformáticos y la lista de genes candidatos extraídos de la minería de literatura, y los dos conjuntos de genes se fusionaron y se utilizaron para identificar redes de interacción proteína-proteína (PPI) y redes reguladoras de genes (GRN). En general, la red reguladora de ceRNA, que consiste en circRNA-lincRNA-lncRNA-miRNA-mRNA, se construyó en base a diversas interacciones para explorar los efectos de los módulos funcionales y los mRNAs, miRNAs, lncRNAs, lincRNAs y circRNAs expresados diferencialmente sobre la fertilidad.
2 Materiales y métodos
Como resumen, el flujo de trabajo general para analizar la recopilación de datos y los métodos para identificar genes clave, vías metabólicas y de señalización, y la construcción de la red reguladora de ceARN lncRNA-miARN-ARNm y los módulos que afectan la alta fertilidad (HF) y la baja fertilidad (LF) en cabras domésticas (Capra hircus) se muestra en la Figura 1.
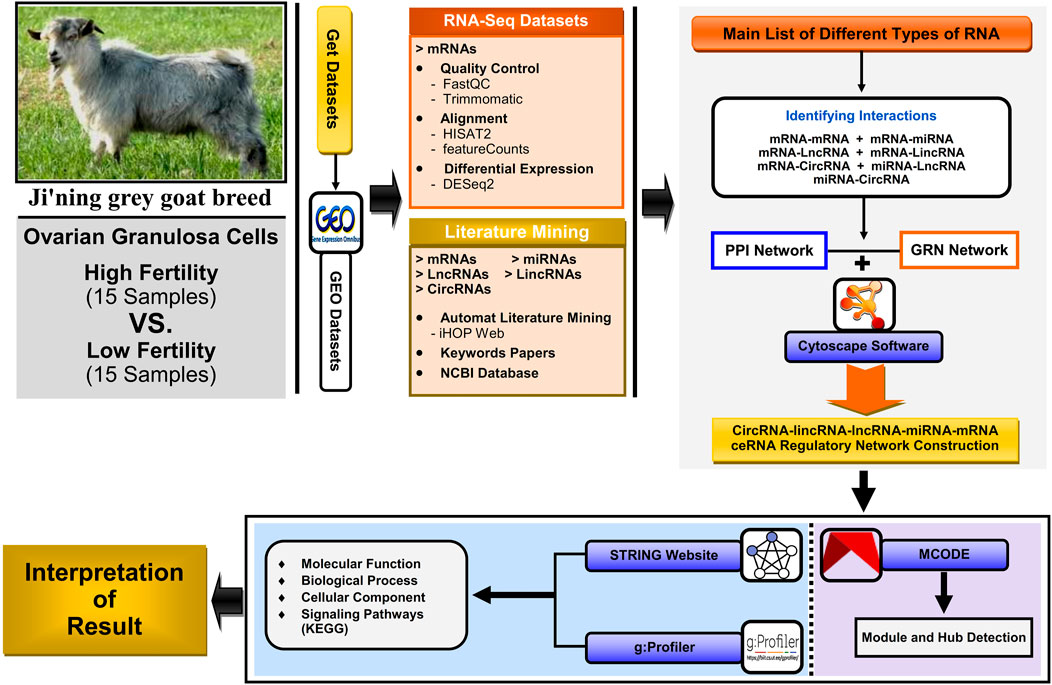 FIGURA 1. Flujo de trabajo para analizar el conjunto de datos scRNA-seq, la minería de literatura, la red de interacción proteína-proteína (PPI), la red reguladora de genes (GRN) y las funciones de ontología aguas abajo, construyendo la red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA; Los análisis de módulos se construyeron y visualizaron utilizando el software Cytoscape v3.9.1.
FIGURA 1. Flujo de trabajo para analizar el conjunto de datos scRNA-seq, la minería de literatura, la red de interacción proteína-proteína (PPI), la red reguladora de genes (GRN) y las funciones de ontología aguas abajo, construyendo la red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA; Los análisis de módulos se construyeron y visualizaron utilizando el software Cytoscape v3.9.1.
2.1 Recopilación de datos
Los datos de scRNA-seq de GC ováricos de cabras Ji’ning Gray de alta y baja fertilidad (HF y LF, respectivamente) se recuperaron de la base de datos pública National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) bajo el número de acceso GSE135897 (www.ncbi.nlm.nih.gov/geo). Este conjunto de datos se produjo utilizando la plataforma GPL15473 Illumina HiSeq 2000 (Li et al., 2021b). Se diseñó un enfoque de estudio de casos y controles para identificar genes expresados diferencialmente (DEG) entre cabras HF y LF. Se analizaron un total de 30 muestras de cabras Ji’ning Gray (primer grupo: 15 con alto tamaño de camada (HF; ≥3 crías) y segundo grupo: 15 con bajo tamaño de camada (LF; ≤2 crías)). La cabra Ji’ning Gray es una raza local y la especie de cabra domesticada más antigua con alta fertilidad en China que tiene estro durante todo el año y un tamaño medio de camada de 2.94 (Huang et al., 2012; Miao et al., 2016a; b). Se recolectaron todos los folículos ováricos de cada grupo de estas cabras grises Ji’ning (desde pequeñas (<3 mm) hasta grandes (˃7 mm)). Luego, los ovocitos y los GC se separaron mecánicamente mediante pipeteo repetido. Se etiquetaron ovocitos de grupos de alta y baja fertilidad. Este estudio se realizó de acuerdo con las directrices de ARRIVE. La preparación de la biblioteca y la secuenciación se realizaron por separado para ovocitos y GC. Se han informado detalles sobre la aprobación de la ética animal, la extracción total de ARN, la preparación de bibliotecas separadas, la secuenciación y la validación de los datos de scRNA-seq (Zhao et al., 2020; Li et al., 2021b).
2.2 Control de calidad y detección de genes expresados diferencialmente
En primer lugar, la calidad de las secuencias de ARN sin procesar se evaluó utilizando el software FastQC v0.11.5 (Andrew, 2010) y el software FastQ Groomer v1.1.5 (Blankenberg et al., 2010); posteriormente, estas secuencias se procesaron previamente con el software Trimmomatic v0.38.0 (Bolger et al., 2014) para eliminar adaptadores, lecturas de baja calidad y cebadores de PCR. Las secuencias de alineación, el mapeo y la identificación de ARN conocidos y nuevos de lecturas se relacionaron con el genoma de referencia de C. hircus (https://ftp.ensembl.org/pub/release-108/fasta/capra_hircus/dna/) utilizando el software HISAT2 v2.2.1 con parámetros predeterminados para determinar el número de lecturas alineadas y no alineadas (Kim et al., 2015). Con respecto a la cuantificación de transcripciones, los recuentos brutos totales de lecturas mapeadas se calcularon utilizando el software featureCounts (v2.0.1) (Liao et al., 2014). Posteriormente, para examinar si la acumulación o degradación de los transcritos estaba relacionada con la fertilidad, las transcripciones y sus niveles de expresión se compararon entre muestras de GC ováricas de cabras Ji’ning Gray de alta y baja fertilidad. Las diferencias en la expresión génica se detectaron a partir de la lectura de recuentos utilizando el software DESeq2 (v2.11.40.7) (Love et al., 2014). El umbral de significación estadística de la expresión diferencial de cada gen se obtuvo con los criterios de log2FC (cambio de pliegue ≥5 y ≤−5) y FDR <0,05 (tasa de descubrimiento falso).
2.3 Minería bibliográfica para identificar genes candidatos para la fertilidad en cabras
Se examinaron varias bases de datos en línea para descubrir circRNAs candidatos, lincRNAs, lncRNAs, miRNAs y mRNAs relevantes para la minería bibliográfica integral. Las bases de datos y documentos de búsqueda en línea incluyeron Google Scholar, PubMed, Web of Science, servicios web iHOP y CrossRef de 2010 a 2023, sin restricciones de idioma. Los términos de búsqueda consistieron en palabras clave y encabezados temáticos específicos de la base de datos para la red reguladora de ceRNA, GC y fertilidad en cabras: razas-cabras; herramientas prácticas-scRNA-seq; y resultado: red de ARNce o ARN reguladores: fertilidad y rasgo de tamaño de camada. Las palabras clave incluyeron cabras, fertilidad, tamaño de la camada, GC ováricas, circRNA, lincRNA, lncRNA, miRNA, mRNA y redes de ceRNA. Para este propósito, primero, los identificadores y sinónimos para cada elemento del marco se fusionaron aplicando el operador booleano «OR». Luego, los elementos del marco se fusionaron aplicando el operador booleano «AND». En total, se identificaron 74 artículos relevantes utilizando bases de datos de búsqueda en línea. Todos los documentos identificados se importaron a Covidence (software de revisión sistemática Covidence, Veritas Health Innovation) y se eliminaron los duplicados. Los artículos incluidos se examinaron adicionalmente en busca de referencias relevantes, y se realizó una verificación de citas. Después de la selección final, se enumeraron 41 artículos con la literatura calificada final, y los ARN reguladores (es decir, circRNAs, lincRNAs, lncRNAs, miRNAs y mRNAs) con diferencias significativas relacionadas con la lista de ARN candidato extraída de la minería de literatura se compilaron como conjuntos de ARN 2-6, respectivamente (Tablas Suplementarias S2-6). Finalmente, el conjunto de ARN 1 (de nuestros análisis bioinformáticos) y los conjuntos de ARN 2-6 (de la minería de literatura) se fusionaron y se utilizaron como archivos de entrada para herramientas de predicción de objetivos, el sitio web STRING y el software Cytoscape para identificar GRN PPI y reconstruir la red y los módulos reguladores de ceARN circRNA-lincRNA-lncRNA-miRNA-ARNm.
2.4 Ontología génica y análisis de enriquecimiento funcional
La anotación de conjuntos de genes y el análisis de enriquecimiento utilizaron DAVID (la base de datos para anotación, visualización y descubrimiento integrado; https://david.ncifcrf.gov/) v6.8 (Sherman y Lempicki, 2009), g:Profiler (https://biit.cs.ut.ee/gprofiler/gost) (Reimand et al., 2016), GeneCards (https://www.genecards.org/) y la base de datos STRING v11.0 (https://string-db.org) (Szklarczyk et al., 2019) para determinar posibles funciones y vías metabólicas. Los genes se asignaron a categorías funcionales utilizando la base de datos Gene Ontology (GO) bajo proceso biológico (BP), función molecular (MF) y componente celular (CC).
2.5 Predicción diana de ARNm expresados diferencialmente y otros tipos de ARN reguladores
La anotación funcional de los tipos de ARN reguladores, es decir, circRNAs, lincRNAs, lncRNAs y miRNAs, consistió en la anotación funcional de sus potenciales genes de ARNm diana. Los genes dirigidos predichos y los tipos de ARN reguladores se predijeron utilizando miRBase (Kozomara et al., 2019) (https://www.mirbase.org/), TargetScan (Grimson et al., 2007), miRanda (http://www.microrna.org/), miRWalk 3.0 (un atlas completo de herramientas de interacción microARN-objetivo que integra 12 herramientas de predicción de miARN-objetivo; http://mirwalk.umm.uni-heidelberg.de/), base de datos NONCODE (Volders et al., 2019) (http://www.noncode.org/), base de datos LNCipedia (Bader et al., 2006) (https://lncipedia.org) y herramienta web CircInteractome (Dudekula et al., 2016) (una herramienta computacional que permite predecir y mapear sitios de unión para RBP y miRNAs en circRNAs reportados; https://circinteractome.nia.nih.gov/). Los genes diana identificados se seleccionaron y se enviaron a las vías DAVID, KEGG, Reactome y la base de datos PANTHER para el enriquecimiento y la validación de genes diana para cada tipo de ARN.
2.6 Construcción de la red reguladora de ceRNA CircRNA-lincRNA-lncRNA-miRNA-mRNA
Basado en la teoría del ceRNA, las funciones globales para todos los RNAs no codificantes pueden servir como una «esponja» endógena para regular circRNAs, lncRNAs, lincRNAs, miRNAs o mRNAs regulados hacia arriba o downregulated circRNAs, lncRNAs, lincRNAs, miRNAs o mRNAs que tienen relaciones inversas entre sí en los pares de interacción mRNA-mRNA, mRNA-miRNA, mRNA-lncRNA y miRNA-circRNA elegidos para construir la red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA (Xu et al., 2019). En este sentido, el análisis de redes PPI se realizó utilizando la base de datos STRING v11.0 (Szklarczyk et al., 2019) (Search Tool for the Retrieval of Interact Genes or Proteins; https://string-db.org), BIND (Biomolecular Interaction Network Database) (Bader et al., 2003), MIPS (Mammalian Protein–Protein Interaction Database) (Pagel et al., 2005) y BioGRID (Biological General Repository for Interaction Datasets) (Chatr-Aryamontri et al., 2012) para explorar las interacciones entre genes en C. hircus. Después de identificar las interacciones entre los tipos de ARN reguladores y los datos de expresión génica (coexpresión), la red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA se reconstruyó y trazó utilizando el software Cytoscape v3.9.1. (Shannon et al., 2003; Instituto Nacional de Ciencias Médicas Generales, Bethesda Softworks, Rockville, MD, EE.UU.). Además, la importancia estadística y topológica de la red se evaluó con el complemento Network Analyzer en el software Cytoscape.
2.7 Agrupación de la red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA y determinación de los principales ARN reguladores del centro
Los módulos o subredes pueden desempeñar un papel importante en la red reguladora principal de ceRNA biológicamente reconstruida, ya que representan un conjunto de nodos con funciones similares que persiguen fines biológicos específicos como módulos funcionales. Para evaluar las propiedades topológicas y los nodos de clúster de la red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA, se utilizaron los complementos de Cytoscape ClusterONE (Shannon et al., 2003) y MCODE (Lotia et al., 2013), algoritmos de agrupamiento para dibujar gráficos direccionales y sin dirección. ClusterONE es un complemento para descubrir subgrafos densamente conectados de una red minimizando los bordes entre clústeres y maximizando los bordes dentro de un clúster. Además, el complemento MCODE se puede utilizar para gráficos dirigidos o no dirigidos. Varios parámetros y la significación estadística y topológica de los módulos reguladores de ceRNA se calcularon utilizando el plugin Cytoscape Network Analyzer v4.4.8 con el valor predeterminado para la red dirigida (Assenov et al., 2008).
3 Resultados y discusión
3.1 Análisis scRNA-seq para identificar genes expresados diferencialmente
En este estudio, investigamos el patrón de los perfiles del transcriptoma de los GC ováricos en cabras. Para proporcionar información sobre la base genética de la fertilidad en cabras Ji’ning Gray, se utilizaron análisis de expresión génica utilizando el conjunto de datos scRNA-seq con el número de acceso GSE135897 (obtenido de la base de datos GEO). El perfil de expresión génica seleccionado tenía 30 muestras, incluidas 15 muestras de GC de cabras de alta fertilidad y 15 muestras de GC de cabras de baja fertilidad. Para realizar este análisis, estas 30 muestras se dividieron en dos grupos (alta y baja fertilidad) para comparar expresiones de perfiles genéticos e identificar DEG significativos. Se identificaron un total de 3.245 genes significativos mediante el procesamiento del perfil de expresión de GC de cabras de alta frente a baja fertilidad. Finalmente, al considerar el umbral de cambio de expresión (cambio de pliegue ≥5 y ≤−5, FDR <0.05), 150 genes se expresaron significativamente diferencialmente en GC de cabras con alta versus baja fertilidad. De estos genes, 80 genes fueron regulados al alza y 70 fueron regulados a la baja (Tabla Suplementaria S1).
Recientemente, se han realizado muchos estudios en genética molecular, bioinformática y sistemas biológicos para descubrir genes candidatos e identificar vías moleculares involucradas en la fertilidad (Ahlawat et al., 2020; An y otros, 2021; Li et al., 2021b). Los genes más identificados estaban relacionados con las funciones reproductivas y el crecimiento folicular ovárico fuertemente regulado y la secreción de hormonas involucradas en la fertilidad; en consecuencia, aumentar o disminuir su expresión en varios momentos puede conducir a PA complejas durante el embarazo (Wang et al., 2019). En este sentido, en un estudio que utilizó el análisis scRNA-seq para tejido ovárico de cabras gestantes y no preñadas, se identificaron cuatro genes (PGR, PRLR, STAR y CYP19A1) que desempeñan un papel importante en la reproducción de cabras (Quan et al., 2019). Además, otro estudio concluyó que algunos lncRNAs en cabras desempeñan un papel clave en la regulación del desarrollo del folículo y el crecimiento celular durante el desarrollo ovárico (Li et al., 2021c).
3.2 Evidencia basada en la minería de literatura para DEG identificados y tipos de ARN reguladores
La minería de la literatura proporcionó evidencia de 81 DEG bien conocidos que no se incluyeron en los resultados investigados por el Tribunal; agregar esta lista de DEG a nuestros DEG descubiertos creó una buena plataforma para análisis posteriores adicionales, incluidos GO y análisis de enriquecimiento funcional. Los DEG identificados basados en la minería de la literatura y su papel en la fertilidad en cabras se recolectaron como conjunto de ARN 2 (Tabla Suplementaria S2). Además, la minería de la literatura proporcionó evidencia para 58, 8, 19 y 55 tipos bien conocidos de ARN reguladores, es decir, circRNAs, lincRNAs, lncRNAs y miRNAs con diferencias significativas, compilados como conjuntos de ARN 3-6, respectivamente (Tablas suplementarias S3-6). La lista de genes en la Tabla Suplementaria S2 se fusionó con la lista de genes en la Tabla Suplementaria S1 y se utilizó para identificar interacciones intergénicas y reconstruir la red PPI. Luego, después de identificar las interacciones entre los tipos de ARN reguladores en los conjuntos de ARN 3-6 juntos y con los datos de expresión génica, se reconstruyó la red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA.
3.3 Ontología génica y análisis de enriquecimiento de vías
La anotación funcional de los términos GO se detectó en base a BP, MF y CC para identificar funciones y vías metabólicas, así como características sistemáticas de la lista combinada de 231 genes DE (conjuntos de ARN 1 y 2), utilizando las bases de datos STRING, DAVID, PANTHER y g:Profiler. Los resultados de la clasificación GO de los DEG para cabras de alta y baja fertilidad se presentan en la Figura 2. Los DEG identificados estuvieron significativamente involucrados en las siguientes funciones: >10 genes de DEG desempeñaron un papel en el proceso celular, la regulación de la PA, la respuesta al estímulo, la respuesta celular al estímulo, la comunicación celular, la regulación del proceso metabólico celular, la organización CC o biogénesis, el proceso reproductivo, la transducción de señales intracelulares, la vía de señalización del receptor beta del factor de crecimiento transformante, la vía de señalización BMP, respuesta a la hormona esteroide y ensamblaje del complejo de proteínas SMAD para PA (Figura 2A). La lista de genes, que incluye genes SMAD2, ESR1, SOX5, BMP4, BMP15, CTNNB1, ERBB2, FGFR1, CDH26, GH, AR y FSHB, estuvo involucrada en la mayoría de los términos de MF que pueden considerarse genes significativos. Además, se identificaron 16 términos MF, como unión iónica, actividad de la proteína quinasa del receptor transmembrana, unión al receptor de señalización, regulador MF, actividad receptor-ligando, unión al factor de crecimiento y actividad del receptor activado beta del factor de crecimiento transformante, que fueron las funciones más significativas asociadas con la fertilidad (Figura 2B). Con respecto a los CC, se identificaron cinco términos GO, a saber, el espacio extracelular, la región de la membrana plasmática, el complejo receptor, la balsa de membrana y la caveola (Figura 2C). Además, el análisis de vías basado en KEGG para DEG identificados se realizó utilizando tres bases de datos en línea, DAVID, STRING y g: Profiler. Los DEG identificados involucrados en la fertilidad se enriquecieron en las vías de señalización que regulan la pluripotencia de las células madre, la interacción citoquina-receptor de citoquinas, la esteroidogénesis ovárica, la meiosis ovocitaria, la maduración de ovocitos mediada por progesterona, la síntesis, secreción y acción de la hormona paratiroidea, la síntesis, secreción y acción de la hormona del crecimiento, la síntesis y secreción de cortisol, la prolactina, TGF-beta, Hippo, MAPK, PI3K-Akt y las vías de señalización FoxO (Figura 3).
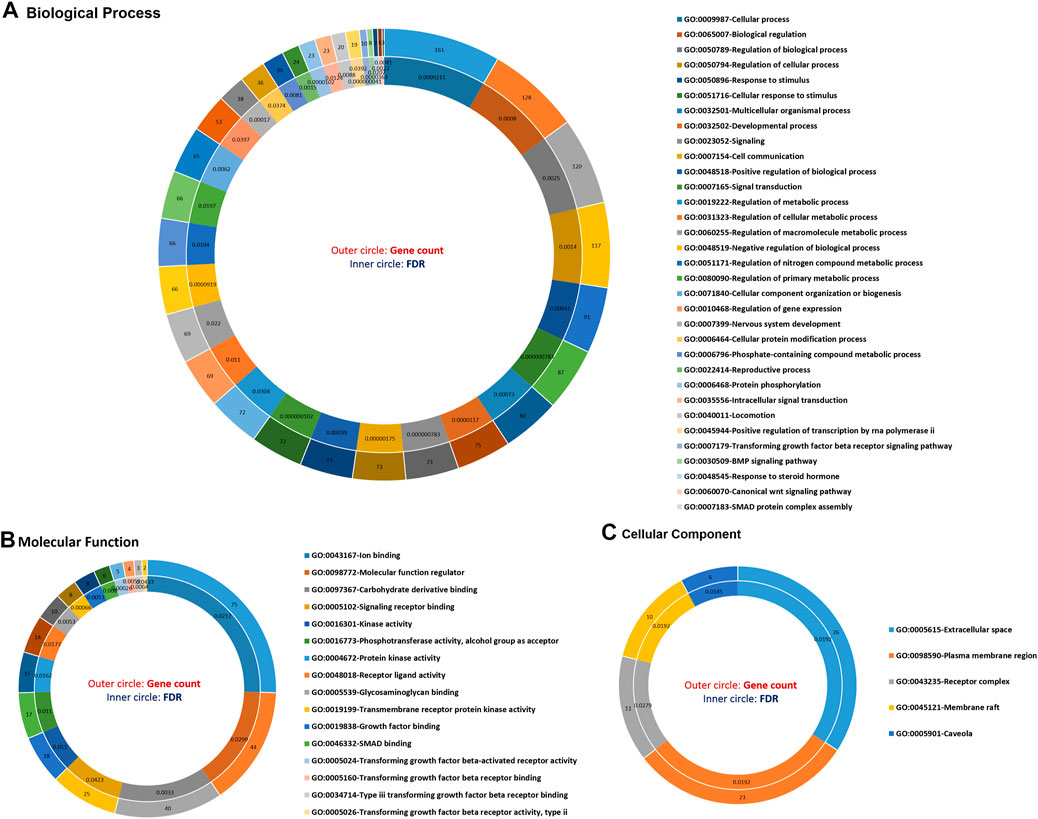 FIGURA 2. Principales términos significativos de ontología génica (GO) enriquecidos utilizando genes expresados diferencialmente asociados con la fertilidad en cabras hembras. (A) Los procesos biológicos significativos, (B) la función molecular significativa, y (C) el componente celular significativo GO términos asociados con la fertilidad en cabras hembras.
FIGURA 2. Principales términos significativos de ontología génica (GO) enriquecidos utilizando genes expresados diferencialmente asociados con la fertilidad en cabras hembras. (A) Los procesos biológicos significativos, (B) la función molecular significativa, y (C) el componente celular significativo GO términos asociados con la fertilidad en cabras hembras.
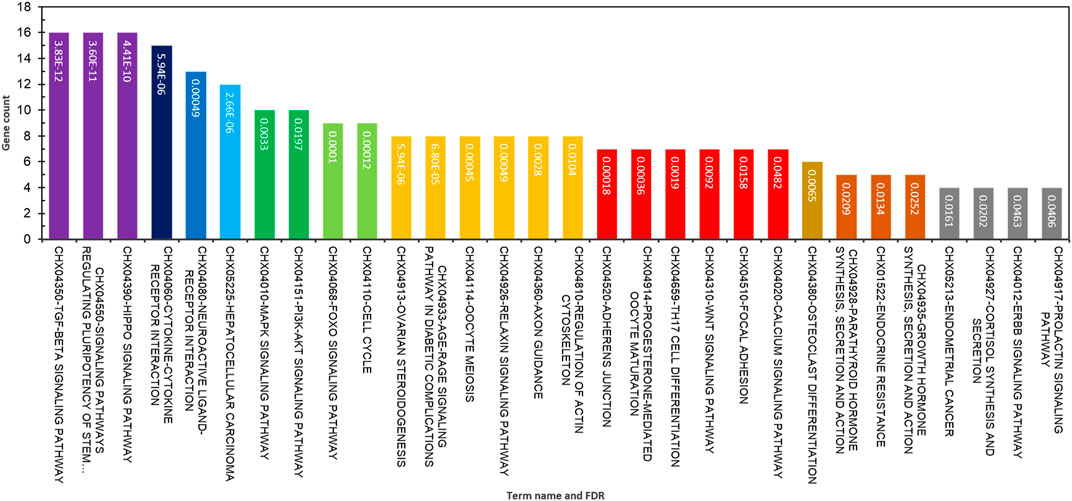 FIGURA 3. Los principales términos significativos de la vía KEGG enriquecidos utilizando genes expresados diferencialmente asociados con la fertilidad en cabras hembras.
FIGURA 3. Los principales términos significativos de la vía KEGG enriquecidos utilizando genes expresados diferencialmente asociados con la fertilidad en cabras hembras.
En cuanto a la función reproductiva femenina, los genes CTNNB1, BMP4, FSHR, TGFB1, BMPR1B y ESR1 se codifican conjuntamente en funciones del proceso del ciclo ovulatorio, desarrollo folicular ovárico, proceso de desarrollo involucrado en la reproducción y diferenciación celular. Por lo tanto, de acuerdo con los genes involucrados en las vías identificadas, ESR1 y BMPR1B se consideraron genes candidatos para las funciones reproductivas, el crecimiento y la diferenciación celular en cabras. Sin embargo, muchos estudios han implicado al gen BMPR1B como uno de los principales genes que controlan la función reproductiva y la fertilidad (por ejemplo, la tasa de ovulación) en pequeños rumiantes, especialmente cabras (Pramod et al., 2013; Ahlawat et al., 2014). Además, el gen FSHR (receptor de hormona foliculoestimulante) desempeña un papel en el crecimiento, la diferenciación y la maduración de los folículos y mejora la función reproductiva en cabras y ovejas (Chen et al., 2017). Además, otro estudio demostró que la FSHR está implicada en la expresión diferencial de los genes receptores de la hormona ARNm ovárico en la fertilidad de las cabras (Saraiva et al., 2011). El gen ESR1 codifica el receptor de estrógeno y el factor de transcripción activado por ligando, y regula los principales genes implicados en el crecimiento, el metabolismo y el embarazo. También se ha informado que la expresión de este gen fue más alta en el riñón, el ovario, el útero y los testículos, pero más baja en el tejido cerebral y cardíaco (Mohammadabadi, 2020). Dado el papel del gen BMP4 en la reproducción, especialmente en el crecimiento y diferenciación de los folículos ováricos y la ovulación, puede considerarse un gen candidato principal para la función reproductiva y la fertilidad (Sharma et al., 2013). El gen CTNNB1 codifica un complejo de proteínas que constituye uniones adherentes (AJs); Son necesarios para la formación y el mantenimiento de las capas celulares epiteliales mediante la regulación del crecimiento celular y la conectividad intercelular. Además, juegan un papel esencial en la reproducción y multiplicación (Zhang et al., 2018b). Por lo tanto, nuestros hallazgos con respecto a los genes FSHR, CTNNB1, BMPR1B y ESR1 fueron consistentes con otros estudios.
3.4 Construcción y agrupamiento de la red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA
Hace una década, Salmena et al. presentaron la hipótesis del ARN endógeno competitivo (Salmena et al., 2011). Las redes reguladoras de ceRNA han proporcionado un nuevo mecanismo de interacción entre los ARN y desempeñan un papel crucial en múltiples PA (Kfir et al., 2018; Yang et al., 2019; Gao et al., 2021). En este sentido, muchos estudios se han dedicado a dilucidar las funciones del ARNce de los ARN no codificantes en algunos rasgos económicamente importantes mediante la construcción de redes de ARN endógeno competitivas (Salmena et al., 2011; Han et al., 2020). Para detectar el mecanismo de cómo los ARN no codificantes (ARNnc) regulan el ARNm a través del miARN esponjoso, se construyó una red reguladora de ARNce con una fusión de pares de interacción ARNm-miARN predichos (mRNA-miRNA), ARNm-lncRNA, ARNm-lincRNA, ARNm-circRNA, miRNA-lncRNA y miRNA-circRNA. La red reguladora de ceRNA reconstruida para mRNAs/genes regulados hacia arriba y hacia abajo y los tipos de RNAs reguladores, que indica conexiones físicas entre dos o más moléculas de proteínas relacionadas con funciones bioquímicas, se presenta en la Figura 4. Sobre la base del conocimiento de las interacciones, esta red reguladora de ceRNA consistió en 310 nodos y 758 bordes y los archivos asociados con las redes se dan en la Tabla Suplementaria S7 (almacenada en formato «.cys» para análisis adicionales). En detalle, se incluyeron en la red 57 circRNAs, 8 lincRNAs, 19 lncRNAs, 51 miRNAs y 175 mRNAs (Figura 4). Como se mencionó, las especies moleculares (circRNAs, lincRNAs, lncRNAs, mRNAs y miRNAs) en redes construidas se indican como nodos y las interacciones entre ellos como bordes. Además, las redes construidas se combinaron en un formato de interacción simple (SIF) utilizando Cytoscape (v3.9.1.) para el análisis topológico. Se evaluaron los parámetros topológicos de la red reguladora y los módulos de ceRNA, como el número de nodos, el número de bordes, el coeficiente de agrupamiento, la longitud de la ruta característica y la densidad de la red para examinar el estado de la comunicación y la transferencia de información de un nodo con otros nodos de redes interactivas, como se presenta en la Tabla 1. En esta red reguladora de ceRNA, 18 genes (SMAD1, SMAD2, SMAD3, SMAD4, TIMP1, ERBB2, BMP15, TGFB1, MAPK3, CTNNB1, BMPR2, AMHR2, TGFBR2, BMP4, ESR1, BMPR1B, AR y TGFB2) tuvieron la mayoría de las interacciones con otros genes de la red. Entre estos 18 genes centrales, cuatro genes (BMP4, BMPR1B, CTNNB1 y ESR1) estaban involucrados en MF y BP, de acuerdo con Pramod et al. (2013), Sharma et al. (2013), Zhang et al. (2018a) y Mohammadabadi (2020). Entre los miRNAs, chi-miR-423-5p, chi-miR-122, chi-miR-187 y chi-miR-133b suprimieron la mayoría de los genes hub identificados como dianas de los miRNAs seleccionados. Además, cuatro de los lincRNAs, es decir, ENSCHIG00000000609, ENSCHIG00000000641, ENSCHIG 00000000886 y ENSCHIG00000002761, interactuaron con los genes TGBFR2, CTNNB1, TGFB2 y SMAD2, respectivamente. Por el contrario, los miRNAs como chi-miR-92a-5p, chi-miR-21-3p, chi-miR-202-3p y chi-miR-223-3p interactuaron con la mayoría de los lncRNAs involucrados en la red reguladora de ceRNAs y se definieron como miRNAs hub que interactuaban con lncRNAs.
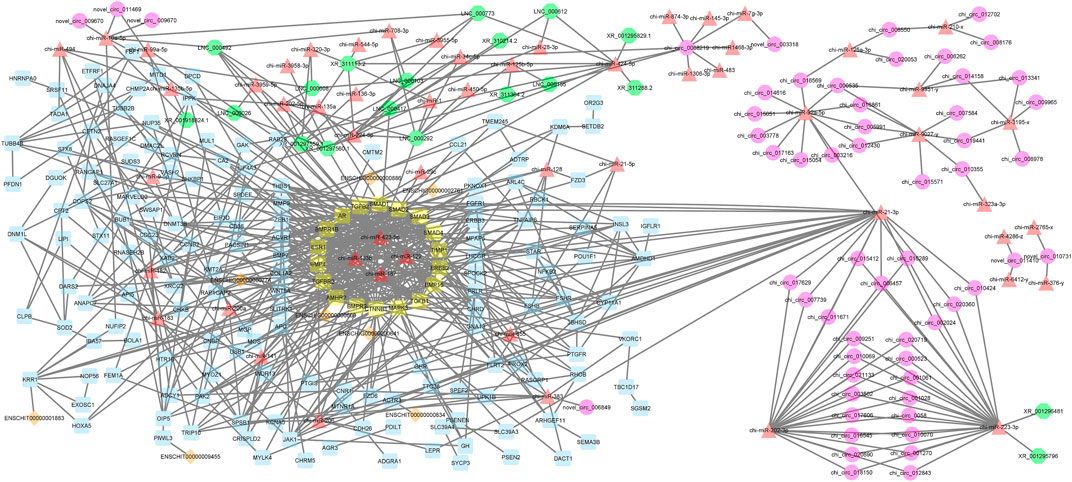 FIGURA 4. red reguladora de ceRNA: se identificaron 57 circRNAs, 8 lincRNAs, 19 lncRNAs, 51 miRNAs y 175 mRNAs en una red interactuada. En esta red, los nodos circulares representan circRNAs, los nodos diamante representan lincRNAs, los nodos octogonales representan lncRNAs, los nodos triangulares representan miRNAs y los nodos cuadriláteros representan mRNAs/genes. Los nodos cuadriláteros amarillos representan los ARNm/genes del centro involucrados en la red. Los bordes negros indican interacciones entre nodos.
FIGURA 4. red reguladora de ceRNA: se identificaron 57 circRNAs, 8 lincRNAs, 19 lncRNAs, 51 miRNAs y 175 mRNAs en una red interactuada. En esta red, los nodos circulares representan circRNAs, los nodos diamante representan lincRNAs, los nodos octogonales representan lncRNAs, los nodos triangulares representan miRNAs y los nodos cuadriláteros representan mRNAs/genes. Los nodos cuadriláteros amarillos representan los ARNm/genes del centro involucrados en la red. Los bordes negros indican interacciones entre nodos.
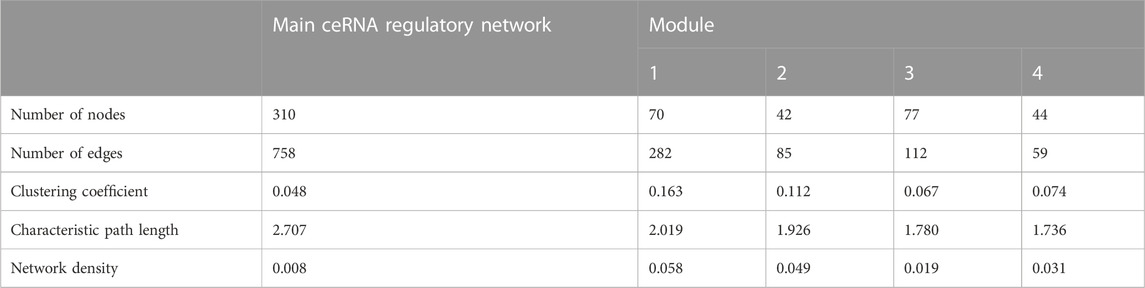 TABLA 1. Estadísticas básicas de red de la red reguladora principal de ceRNA generada y sus módulos.
TABLA 1. Estadísticas básicas de red de la red reguladora principal de ceRNA generada y sus módulos.
En este estudio, la agrupación de la red reguladora circRNA-lincRNA -lncRNA-miRNA-mRNA se realizó utilizando los complementos ClusterONE (Shannon et al., 2003) y MCODE (Lotia et al., 2013), de acuerdo con los algoritmos de agrupamiento utilizados para determinar subredes o módulos significativos mediante un enfoque integrado. Había cuatro módulos candidatos involucrados en la fertilidad de las cabras; las interacciones de nodo de cada componente se describen en la Tabla Suplementaria S7.
Curiosamente, el módulo 1 consistía en 70 nodos y 282 bordes y, en detalle, comprendía 2 lincRNAs, 12 miRNAs y 56 mRNAs. En este módulo, SMAD1, SMAD2, SMAD3, SMAD4, TGFBR2, CTNNB1 y ESR1 eran genes hub. Además, chi-miR-128, chi-miR-122, chi-miR-187, chi-miR-200a, chi-miR-206 y chi-miR-133b, como miRNAs hub, suprimieron la mayoría de los genes implicados como DNMT3B, SMAD2, SMAD4, CHRD, RBCK1, BMPR1B, KDM6A, KRR1, GNA13, ERBB3, TGFB2, ERBB2, SPSB1, TGFBR2, BMPR2, CCL21, THBS1, PRLR, ESR1 y RASGRP1. Además, en este módulo, los lincRNAs ENSCHIG00000000609 y ENSCHIG00000000886 interactuaron con TGFBR2 y TGFB2, respectivamente. Todos los genes hub-hub en este módulo estaban involucrados en las vías de señalización metabólica que se analizaron (Figura 5).
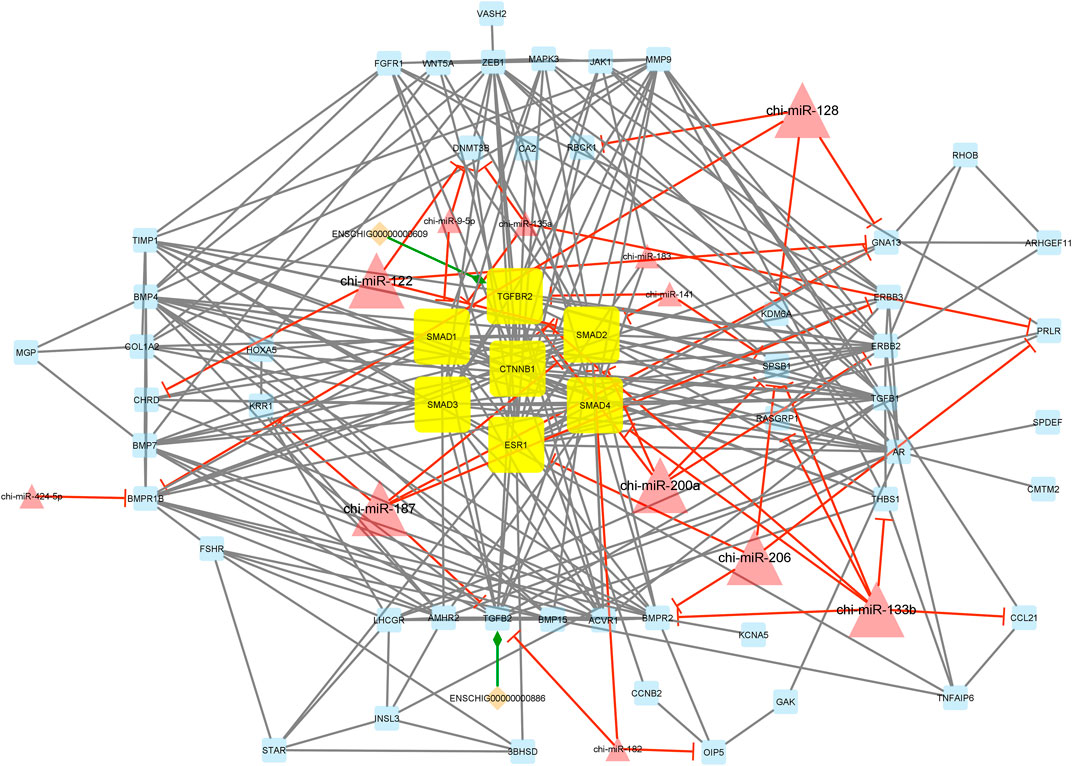 FIGURA 5. Módulo 1: Se identificaron 2 lincRNAs, 12 miRNAs y 56 mRNAs en una red interactuada. En esta red, los nodos de diamante representan lincRNAs, los nodos triangulares representan miRNAs y los nodos cuadriláteros representan mRNAs/genes. Los nodos cuadrilátero amarillo y triángulo grande representan ARNm/genes centrales y miARN involucrados en la red, respectivamente. Los bordes indican las interacciones; los bordes negros representan las interacciones ARNm-ARNm, los bordes verdes representan las interacciones lincRNA-ARNm, y los bordes rojos representan las interacciones miRNA-ARNm.
FIGURA 5. Módulo 1: Se identificaron 2 lincRNAs, 12 miRNAs y 56 mRNAs en una red interactuada. En esta red, los nodos de diamante representan lincRNAs, los nodos triangulares representan miRNAs y los nodos cuadriláteros representan mRNAs/genes. Los nodos cuadrilátero amarillo y triángulo grande representan ARNm/genes centrales y miARN involucrados en la red, respectivamente. Los bordes indican las interacciones; los bordes negros representan las interacciones ARNm-ARNm, los bordes verdes representan las interacciones lincRNA-ARNm, y los bordes rojos representan las interacciones miRNA-ARNm.
Además, el módulo 2 consistió en 42 nodos y 85 bordes y, en detalle, comprendió 2 lincRNAs, 8 miRNAs y 32 mRNAs. En este módulo, SMAD2, SMAD4, CTNNB1 y TIMP1 eran genes centrales. Además, chi-miR-182, chi-miR-200a, chi-miR-187 y chi-miR-122, como miRNAs de concentrador, suprimieron la mayoría de los genes como SMAD2, TRIP10, COPS2, PAK2, KMT2A, ERBB2, APC, GNA13, USB1 y FZD6. Entre estos, el gen SMAD2 fue suprimido más que otros genes involucrados en la red. Además, en este módulo, los lincRNAs ENSCHIG00000000641 y ENSCHIG00000002761 interactuaron con CTNNB1 y SMAD2, respectivamente (Figura 6).
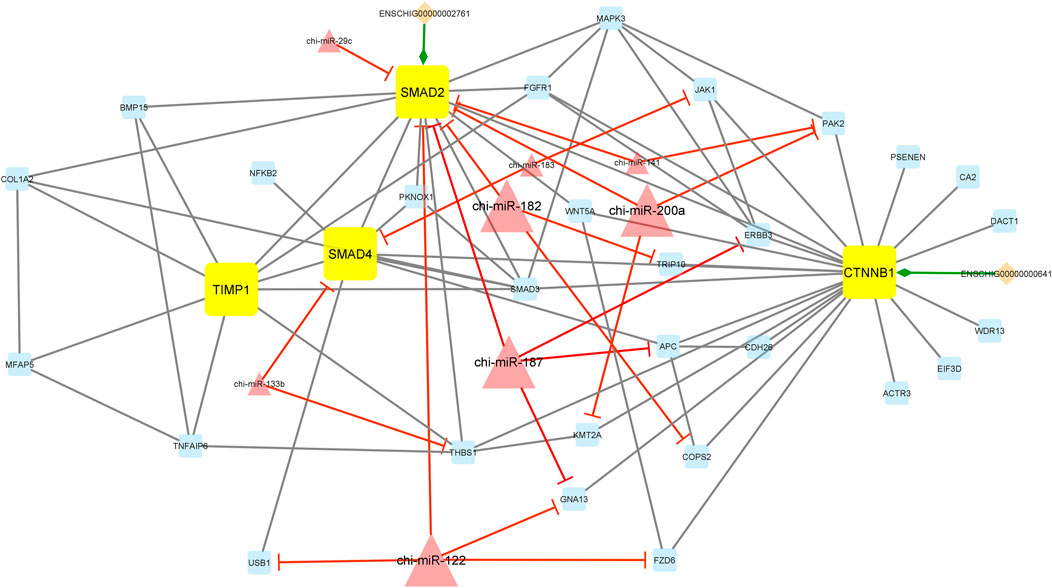 FIGURA 6. Módulo 2: Se identificaron 2 lincRNAs, 8 miRNAs y 32 mRNAs en una red interactuada. En esta red, los nodos de diamante representan lincRNAs, los nodos triangulares representan miRNAs y los nodos cuadriláteros representan mRNAs/genes. Los nodos cuadrilátero amarillo y triángulo grande representan ARNm/genes centrales y miARN involucrados en la red, respectivamente. Los bordes indican las interacciones; los bordes negros representan las interacciones ARNm-ARNm, los bordes verdes representan las interacciones lincRNA-ARNm, y los bordes rojos representan las interacciones miRNA-ARNm.
FIGURA 6. Módulo 2: Se identificaron 2 lincRNAs, 8 miRNAs y 32 mRNAs en una red interactuada. En esta red, los nodos de diamante representan lincRNAs, los nodos triangulares representan miRNAs y los nodos cuadriláteros representan mRNAs/genes. Los nodos cuadrilátero amarillo y triángulo grande representan ARNm/genes centrales y miARN involucrados en la red, respectivamente. Los bordes indican las interacciones; los bordes negros representan las interacciones ARNm-ARNm, los bordes verdes representan las interacciones lincRNA-ARNm, y los bordes rojos representan las interacciones miRNA-ARNm.
Además, el módulo 3 consistía en 77 nodos y 112 aristas y, en detalle, comprendía 1 circRNA, 7 lncRNAs, 16 miRNAs y 53 mRNAs. En este módulo, TUBB4B, CETN2, XAB2 y 3BHSD tuvieron la mayor interacción con otros genes del módulo como genes centrales. Además, chi-miR-200a, chi-miR-494 y chi-miR-128, como miRNAs centrales, suprimieron los genes ANAPC7, KMT2A, PAK2, MARVELD2, SLC27A1, TADA1, FZD3, KDM6A, FSHB y PKNOX1. Entre estos, chi-miR-128 miRNA suprimió la mayoría de los genes involucrados en la red. Además, en este módulo, XR_001297559.1 y XR_001297560.1 circRNAs interactuaron con la mayoría de los miRNAs como chi-miR-494, chi-miR-3959-5p, chi-miR-494, chi-miR-1, chi-miR-202-5p, chi-miR-34c-5p, chi-miR-320-3p y chi-miR-136-3p (Figura 7).
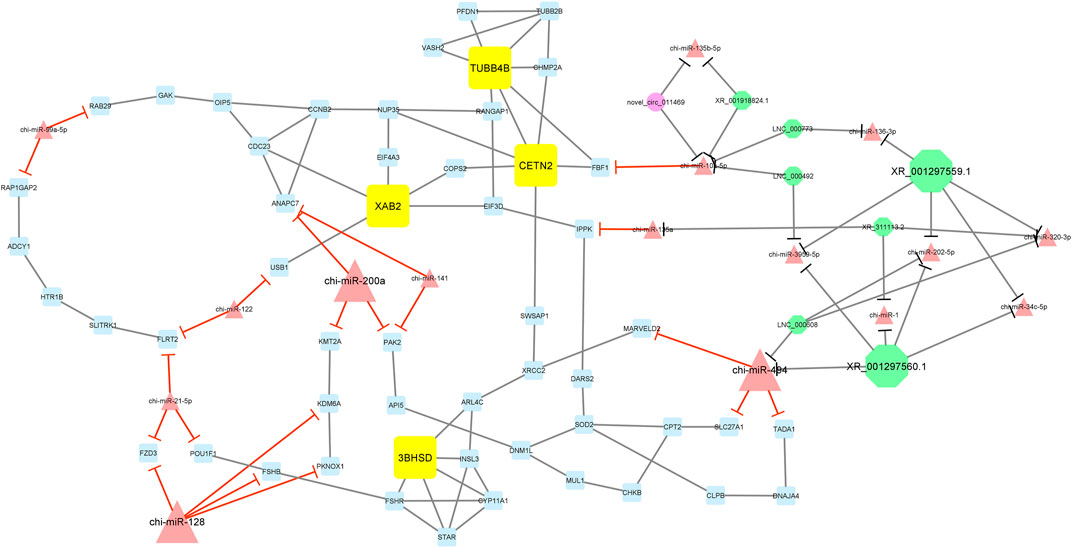 FIGURA 7. Módulo 3: Se identificaron 1 circRNA, 7 lncRNAs, 16 miRNAs y 53 mRNAs en una red interactuada. En esta red, los nodos circulares representan circRNAs, los nodos octogonales representan lncRNAs, los nodos triangulares representan miRNAs y los nodos cuadriláteros representan mRNAs/genes. Los nodos cuadrilátero amarillo, triángulo grande y octogonal grande representan ARNm/genes centrales, miARN y lncRNAs involucrados en la red, respectivamente. Los bordes indican interacciones; los bordes negros representan las interacciones ARNm-ARNm, lncRNA-miARN y circARN-miARN, y los bordes rojos representan las interacciones miARN-ARNm.
FIGURA 7. Módulo 3: Se identificaron 1 circRNA, 7 lncRNAs, 16 miRNAs y 53 mRNAs en una red interactuada. En esta red, los nodos circulares representan circRNAs, los nodos octogonales representan lncRNAs, los nodos triangulares representan miRNAs y los nodos cuadriláteros representan mRNAs/genes. Los nodos cuadrilátero amarillo, triángulo grande y octogonal grande representan ARNm/genes centrales, miARN y lncRNAs involucrados en la red, respectivamente. Los bordes indican interacciones; los bordes negros representan las interacciones ARNm-ARNm, lncRNA-miARN y circARN-miARN, y los bordes rojos representan las interacciones miARN-ARNm.
Finalmente, el módulo 4 consistió en 44 nodos y 59 bordes y, en detalle, comprendió 5 lncRNAs, 2 miRNAs y 37 mRNAs. En este módulo, MAPK3, JAK1 y ERBB3 tuvieron la mayor interacción con otros genes del módulo como genes centrales. Además, casi todos los genes involucrados en este módulo fueron suprimidos por miRNAs chi-miR-423-5p y chi-miR-187. Además, el miARN chi-miR-423-5p interactuó con los cinco lncRNAs, es decir, XR_001297560.1, XR_001297559.1, LNC_000292, LNC_000417 y LNC_000492, involucrados en el módulo (Figura 8).
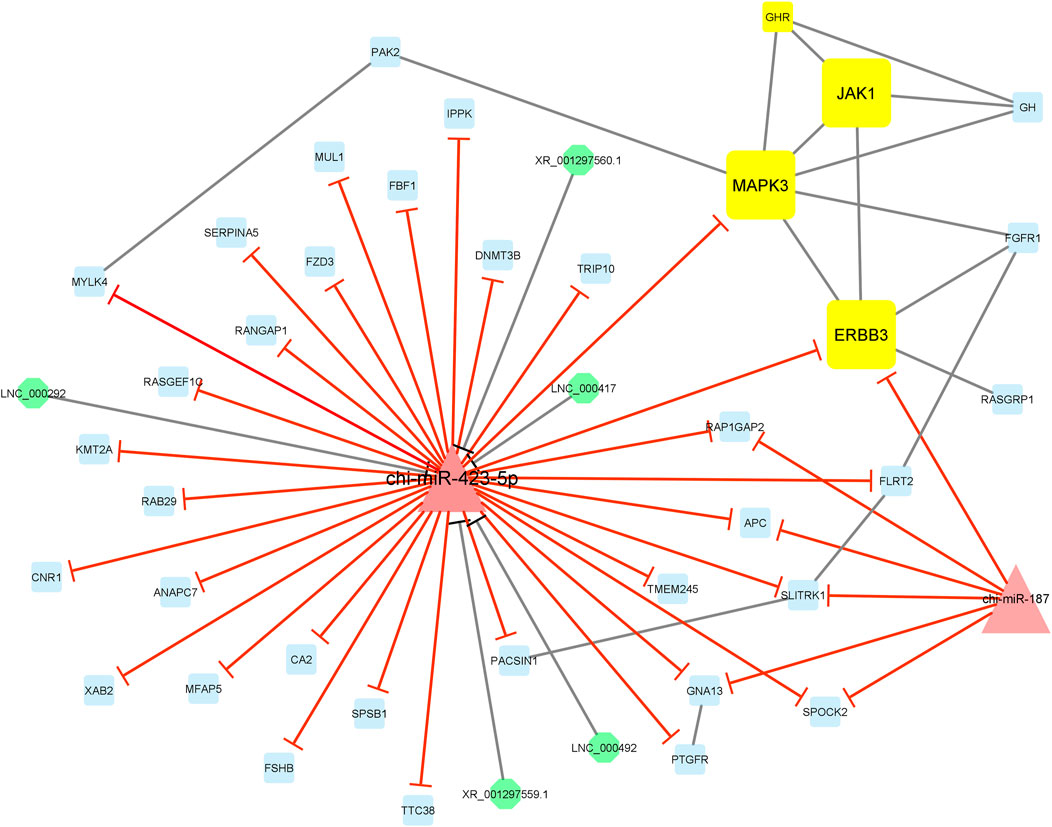 FIGURA 8. Módulo 4: Se identificaron 5 lncRNAs, 2 miRNAs y 37 mRNAs en una red interactuada. En esta red, los nodos octogonales representan lncRNAs, los nodos triangulares representan miRNAs y los nodos cuadriláteros representan mRNAs/genes. Los nodos cuadrilátero amarillo y triángulo grande representan ARNm/genes centrales y miARN involucrados en la red, respectivamente. Los bordes indican interacciones; los bordes negros representan las interacciones ARNm-ARNm y lncRNA-miARN, y los bordes rojos representan las interacciones miARN-ARNm.
FIGURA 8. Módulo 4: Se identificaron 5 lncRNAs, 2 miRNAs y 37 mRNAs en una red interactuada. En esta red, los nodos octogonales representan lncRNAs, los nodos triangulares representan miRNAs y los nodos cuadriláteros representan mRNAs/genes. Los nodos cuadrilátero amarillo y triángulo grande representan ARNm/genes centrales y miARN involucrados en la red, respectivamente. Los bordes indican interacciones; los bordes negros representan las interacciones ARNm-ARNm y lncRNA-miARN, y los bordes rojos representan las interacciones miARN-ARNm.
Según la literatura, los genes relacionados con la función reproductiva en cabras lecheras, incluidos los genes CCNB2, AR, ADCY1, DNMT3B, SMAD2, AMHR2, ERBB2 y FGFR1, se seleccionaron específicamente en cabras con alta fertilidad (Lai et al., 2016; Zonaed Siddiki et al., 2020). Por lo tanto, nuestros resultados proporcionaron evidencia adicional de la asociación entre nueve genes hub (AR, SMAD1, SMAD2, SMAD3, SMAD4, AMHR2, TGFBR2, CTNNB1 y ERBB2) y las funciones reproductivas femeninas en cabras. Se informó que el gen ERBB2 (Erb-b2 receptor tirosina quinasa 2) es un receptor de hormona esteroide implicado en la señalización del calcio (Zwick et al., 1999). Del mismo modo, el gen AR (receptor de andrógenos) es una proteína que implica un factor de transcripción de unión al ADN y la unión a la cromatina; además, desempeña un papel clave en la reproducción mediante la transmisión de señales de andrógenos (Zonaed Siddiki et al., 2020). TGFBR2 (receptor beta 2 del factor de crecimiento transformante) es un gen codificador de proteínas que desempeña un papel importante en la señalización del receptor TGF-beta, la activación de SMAD, la actividad de la transferasa, la transferencia de grupos que contienen fósforo y la actividad de la proteína tirosina quinasa (Yao et al., 2022). Los genes CTNNB1 (catenina beta 1) y TGF-β2 se asociaron con el factor de crecimiento transformante beta (TGF-β), vías de señalización que regulan la pluripotencia de las células madre, infección por HTLV-I, interacción neuroactiva ligando-receptor, Wnt y vías de señalización de hipopótamo (Li et al., 2021b). Los genes SMAD2, SMAD3 y SMAD4 desempeñan un papel crítico en el crecimiento y la diferenciación de las células ováricas, en consonancia con algunos aspectos de la ovulación (Li et al., 2008; Fortin et al., 2014).
En particular, el gen AMHR2 (receptor hormonal antimulleriano tipo 2) se asocia con la actividad de la transferasa, el transporte de grupos que contienen fósforo y la actividad de la proteína tirosina quinasa. Este gen también está implicado en el crecimiento y desarrollo de los folículos ováricos en bovinos y caprinos (Monniaux et al., 2011). Además, los genes BMP15 y GDF9, como genes candidatos, son miembros de la familia del factor de crecimiento beta (TGF-β), directamente relacionados con el hermanamiento, el aumento de la ovulación y el crecimiento y desarrollo de folículos ováricos en ovejas y cabras (Pramod et al., 2013). El gen FOXL2 está implicado en el crecimiento y la función ovárica, así como en las primeras etapas del crecimiento ovárico de los mamíferos (Baron et al., 2005). FSHB (subunidad beta de la hormona foliculoestimulante) es un gen crítico en la actividad de la hormona foliculoestimulante y el metabolismo de la hormona peptídica. Además, las variaciones en este gen pueden afectar la señalización de la diferenciación del folículo y la ovulación (Zi et al., 2020). El gen GH (hormona del crecimiento) está directamente involucrado en los cambios inducidos por la nutrición en el control de las funciones reproductivas, por ejemplo, el crecimiento y desarrollo folicular ovárico, la división celular y la ovulación (Zhang et al., 2011). El gen PRLR (receptor de prolactina) se ha detectado en varios tejidos, incluyendo el cerebro, el ovario, la placenta y el útero en varios mamíferos, especialmente pequeños rumiantes. Esta hormona está involucrada en muchas actividades endocrinas y es esencial para la función reproductiva (Ozmen et al., 2011). Por lo tanto, de acuerdo con las funciones de los genes hub identificados en la red y los módulos reguladores circRNA-lincRNA-lncRNA-miRNA-mRNA, concluimos que estos genes desempeñan un papel importante en el rendimiento reproductivo y la fertilidad de las cabras. En este sentido, están involucrados en las glándulas endocrinas, el crecimiento, la diferenciación celular, así como la maduración del folículo y el aumento de la ovulación y podrían seleccionarse en programas de reproducción para aumentar los beneficios económicos. Además, la mayoría de los genes implicados en la red reguladora de ceRNA codifican vías de señalización que regulan la pluripotencia de las células madre, la interacción citocina-receptor de citoquinas, la esteroidogénesis ovárica y la interacción ligando-receptor neuroactivo, confirmando así las funciones de los genes involucrados, especialmente los genes centrales.
Las vías metabólicas y de señalización codificadas por los genes implicados en la red reguladora circRNA-lincRNA-lncRNA-miRNA-mRNA y los módulos se presentan en las Figuras 2, 3. Las vías de señalización de TGF-beta, que regulan la pluripotencia de las células madre, Hippo, MAPK, PI3K-Akt y FoxO están codificadas por la red y los módulos reguladores de ceRNA. La vía de señalización TGF-beta codifica un gran grupo de proteínas estructurales relacionadas, incluidas las proteínas morfogenéticas óseas, el factor de crecimiento y la diferenciación (Liu et al., 2018). Las vías de señalización que regulan la pluripotencia de las células madre están codificadas por células madre pluripotentes (PSC), que muestran potencial para producir las tres capas de células germinales. Cabe destacar que las células madre embrionarias (ESC) se derivan de la masa celular interna (MCI) de embriones en estadio de blastocisto (Mossahebi-Mohammadi et al., 2020). La vía de señalización del hipopótamo está implicada en la inhibición de la proliferación celular y en la mejora de la apoptosis (Saucedo y Edgar, 2007). MAPK3 (mitogen-activated protein kinase 3) es un gen de la familia MAP quinasa. Este gen desempeña un papel importante en la cascada de señalización que regula diversos procesos celulares como la proliferación, la diferenciación y la progresión del ciclo celular en respuesta a una variedad de señales extracelulares (Miao et al., 2016a). Las vías metabólicas funcionales, como la interacción citocina-receptor de citoquinas, la esteroidogénesis ovárica, la interacción neuroactiva ligando-receptor y la secreción y acción de la síntesis de la hormona del crecimiento también están codificadas en estas subredes, con papeles importantes en la reproducción y la fertilidad.
En este estudio, se realizó un enfoque computacional con una red reguladora de circRNA-lincRNA-lncRNA-miRNA-mRNA utilizando perfiles de expresión predichos y validados de ARN. La expresión diferencial espaciotemporal en varios tejidos, especialmente los folículos ováricos, cubrió los roles potenciales de los tipos de ARN en la regulación transcripcional y posttranscripcional de los genes involucrados en la fertilidad. Una explicación común para las inconsistencias en nuestros resultados fueron las diferencias en las técnicas moleculares aplicadas (GWAS, Microarray, scRNA-seq y expresión génica relativa simple), las diferencias en el tejido ovárico, el tiempo de muestreo y los algoritmos bioinformáticos. Las limitaciones de los estudios actuales incluyen la falta de un conjunto de datos único y completo con condiciones ambientales similares y tejido ovárico de razas de cabras similares.
En resumen, concluimos que las firmas transcriptómicas identificadas son biomarcadores potencialmente importantes para comprender mejor las vías funcionales involucradas en la fertilidad en cabras hembras. Se necesitan más esfuerzos para dilucidar los tipos funcionales biológicos específicos de ARN en la reproducción y la fertilidad. Además, nuestros hallazgos integraron circRNAs, lincRNAs, lncRNAs, miRNAs y mRNAs basados en un enfoque integrado de análisis bioinformáticos y minería de literatura para construir redes reguladoras de ceRNA. Aunque esto puede considerarse un enfoque sólido para detectar información significativa sobre los BP, se necesitará más investigación para confirmar nuestros resultados.
4 Conclusión
Este estudio utilizó un enfoque novedoso para combinar varios tipos de ARN como una red integrada en la fertilidad de las cabras. Los análisis de los datos de scRNA-seq dieron como resultado la identificación de 150 DEG en cabras con fertilidad alta versus baja. Entre ellos, 80 genes fueron regulados al alza y 70 fueron regulados a la baja. Además, se obtuvieron 81 ARNm/genes, 58 circRNAs, 8 lincRNAs, 19 lncRNAs y 55 miRNAs, todos tipos bien conocidos de RNAs reguladores, de la minería bibliográfica. Utilizando circRNA-lincRNA-lncRNA-miRNA-mRNA ceRNA, se construyó una red reguladora y estos ARN identificados se asociaron principalmente con actividades reguladoras transcripcionales y actividades de unión a receptores de señalización en términos de MF, así como funciones reproductivas como el ciclo de ovulación, el desarrollo del folículo ovárico, el crecimiento y las células de diferenciación basadas en BP. Además, nuestros resultados son un recurso valioso para dilucidar las redes moleculares y las funciones de los DEG subyacentes al desarrollo folicular ovárico, y aumentan la comprensión de la base genética de las cabras de alta frente a baja fertilidad.
Declaración de disponibilidad de datos
Los conjuntos de datos presentados en este estudio se pueden encontrar en repositorios en línea. Los nombres del repositorio / repositorios y el (los) número (s) de acceso se pueden encontrar en el artículo / Material complementario.
Declaración ética
El estudio en animales fue revisado y aprobado por el Parque Científico de la Universidad de Teherán.
Contribuciones del autor
Conceptualización: FG, MS, MN y AB; curación de datos: FG, VD y SG; análisis e investigación formales: FG; metodología: FG, VD y AB; redacción: preparación del borrador original: FG, MN, AJ y AB; redacción: revisión y edición: MS, SM-A, JK, RA-A y HB; supervisión: MS y SM-A. Todos los autores contribuyeron al artículo y aprobaron la versión presentada.
Reconocimientos
Los autores expresan su gratitud por la base de datos de repositorio abierto Gene Expression Omnibus (GEO) por proporcionar una plataforma para acceder a datos moleculares.
Conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un posible conflicto de intereses.
Nota del editor
Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo, o reclamo que pueda ser hecho por su fabricante, no está garantizado ni respaldado por el editor.
Material complementario
El material complementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fgene.2023.1195480/full#supplementary-material
Referencias
Ahlawat, S., Sharma, R., Maitra, A., Tantia, M., Roy, M. y Mandakmale, S. (2014). Nuevos polimorfismos genéticos en cabra india en el gen BMPR1B. Indio J. Anim. 84, 37–42.
Ahlawat, S., Sharma, R., Roy, M., Mandakmale, S., Prakash, V. y Tantia, M. (2016). Genotipado de nuevos SNP en los genes BMPR1B, BMP15 y GDF9 para la asociación con la prolificidad en siete razas de cabras indias. Anim. Biotechnol. 27, 199–207. doi:10.1080/10495398.2016.1167706
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Ahlawat, S., Arora, R., Sharma, R., Sharma, U., Kaur, M., Kumar, A., et al. (2020). El perfil del transcriptoma cutáneo de las cabras Changthangi destaca la relevancia de los genes involucrados en la producción de Pastina. 10, 1–10. doi:10.1038/s41598-020-63023-6
Resumen de PubMed | Texto completo de CrossRef | Google Académico
An, X., Zhang, Y., Li, F., Wang, Z., Yang, S. y Cao, B. (2021). Análisis del transcriptoma completo: Implicación en la regulación del ciclo estral. Biología 10 (6), 464. doi:10.3390/biology10060464
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Andrés, S. (2010). Una herramienta de control de calidad para datos de secuencia de alto rendimiento. Control de calidad rápido. 532, 1. Disponible en línea en: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Assenov, Y., Ramírez, F., Schelhorn, S. E., Lengauer, T. y Albrecht, M. (2008). Cálculo de parámetros topológicos de redes biológicas. Bioinformática 24 (2), 282–284. doi:10.1093/bioinformatics/btm554
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Bader, G. D., Betel, D. y Hogue, C. W. (2003). Bind: La base de datos de la red de interacción biomolecular. Ácidos nucleicos res. 31 (1), 248–250. doi:10.1093/nar/gkg056
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Bader, G. D., Cary, M. P. y Sander, C. (2006). Guía de ruta: una lista de recursos de ruta. Ácidos nucleicos res. 34 (1), D504–D506. doi:10.1093/nar/gkj126
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Bahrami, A., Miraie-Ashtiani, S. R., Sadeghi, M. y Najafi, A. (2017). red miRNA-mRNA implicada en el interactoma foliculogénesis: enfoque de biología de sistemas. Reproducción 154, 51–65. doi:10.1530/REP-17-0049
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Baron, D., Batista, F., Chaffaux, S., Cocquet, J., Cotinot, C., Cribiu, E., et al. (2005). El gen Foxl2 y el desarrollo del ovario: Una historia sobre cabra, ratón, pez y mujer. Reprod. Nutr. Dev. 45, 377–382. doi:10.1051/rnd:2005028
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Blankenberg, D., Gordon, A., Von Kuster, G., Coraor, N., Taylor, J., Nekrutenko, A., et al. (2010). Manipulación de datos FASTQ con galaxy. Bioinformática 26, 1783-1785. doi:10.1093/bioinformatics/btq281
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Bolger, A. M., Lohse, M. y Usadel, B. (2014). Trimmomatic: Un recortador flexible para los datos de secuencia de Illumina. Bioinformática 30, 2114–2120. doi:10.1093/bioinformática/btu170
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Bugrim, A., Nikolskaya, T. y Nikolsky, Y. (2004). Predicción temprana del metabolismo y la toxicidad de los fármacos: enfoque y modelado de la biología de sistemas. Drogas Discov. Hoy 9 (3), 127–135. doi:10.1016/S1359-6446(03)02971-4
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Chatr-Aryamontri, A., Breitkreutz, B. J., Heinicke, S., Boucher, L., Winter, A., Stark, C., et al. (2012). La base de datos de interacción BioGRID: actualización de 2013. Ácidos nucleicos res. 41 (D1), D816–D823. doi:10.1093/nar/gks1158
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Chen, H. Y., Shen, H., Jia, B., Zhang, Y. S., Wang, X. H. y Zeng, X. C. (2015). Expresión génica diferencial en ovarios de ovejas negras Qira y ovejas Hetman utilizando la técnica RNA-Seq. PloS One 10, e0120170. doi:10.1371/journal.pone.0120170
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Chen, X., Long, W., Sun, Z., Feng, W., Li, P. y Xu, H. (2017). Estudio de correlación entre el gen FSHR y los rasgos reproductivos de la cabra nativa de Guizhou (Capra hircus). J. Agric. Biotechnol. 25, 94–101. doi:10.3969/j.issn.1674-7968.2017.02.015
Choi, Y. H. y Kim, J. K. (2019). Disección de la heterogeneidad celular mediante secuenciación de ARN unicelular. Mol. Células. 42, 189–199. doi:10.14348/molcells.2019.2446
Resumen de PubMed | Texto completo de CrossRef | Google Académico
de Lima, L. G., de Souza, N. O. B., Rios, R. R., de Melo, B. A., dos Santos, L. T. A., Silva, K. D. M., et al. (2020). Avances en técnicas de genética molecular aplicadas a la selección por tamaño de camada en cabras (Capra hircus): Una revisión. J. Appl. Anim. 48, 38–44. doi:10.1080/09712119.2020.1717497
Deniz, E. y Erman, B. (2017). ARN largo no codificante (lincRNA), un nuevo paradigma en el control de la expresión génica. Funct. Integrar. Genom. 17, 135–143. doi:10.1007/s10142-016-0524-x
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Dudekula, D. B., Panda, A. C., Grammatikakis, I., De, S., Abdelmohsen, K. y Gorospe, M. (2016). CircInteractome: Una herramienta web para explorar ARN circulares y sus proteínas y microARN que interactúan. ARN Biol. 13 (1), 34–42. doi:10.1080/15476286.2015.1128065
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Fatet, A., Pellicer-Rubio, M-T. y Leboeuf, B. (2011). Ciclo reproductivo de las cabras. Anim. Reprod. 124, 211–219. doi:10.1016/j.anireprosci.2010.08.029
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Fortin, J., Boehm, U., Deng, C. X., Treier, M. y Bernard, D. J. (2014). La síntesis de la hormona foliculoestimulante y la fertilidad dependen de SMAD4 y FOXL2. FASEB J. 28 (8), 3396–3410. doi:10.1096/fj.14-249532
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Gao, L., Zhao, Y., Ma, X. y Zhang, L. (2021). Análisis integrado de la red lncRNA-miRNA-mRNA ceRNA y los posibles indicadores de pronóstico en sarcomas. BMC Med. Genet. 14, 67–11. doi:10.1186/s12920-021-00918-x
Ghafouri, F., Bahrami, A., Sadeghi, M., Miraei-Ashtiani, S. R., Bakherad, M., Barkema, H. W., et al. (2021). Las redes multicapa ómicas proporcionan nuevos conocimientos mecanicistas y funcionales sobre el almacenamiento de grasa y el metabolismo de los lípidos en las aves de corral. Frente. Jineta. 12, 646297. doi:10.3389/fgene.2021.646297
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Grimson, A., Farh, K. K. H., Johnston, W. K., Garrett-Engele, P., Lim, L. P. y Bartel, D. P. (2007). Especificidad dirigida a microARN en mamíferos: determinantes más allá del apareamiento de semillas. Mol. 27 (1), 91–105. doi:10.1016/j.molcel.2007.06.017
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Hallock, P., y Thomas, M. A. (2012). Integración del proteoma y transcriptoma de la enfermedad de Alzheimer: Un modelo de red integral de una enfermedad compleja. OMICS J. Integr. Biol. 16 (1-2), 37–49. doi:10.1089/omi.2011.0054
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Han, R., Han, L., Wang, S. y Li, H. (2020). El análisis del transcriptoma completo del tejido mesenquima en astas de ciervo sika reveló la red reguladora de CeRNAs asociada con el desarrollo de la cornamenta. Frente. Jineta. 10, 1403. doi:10.3389/fgene.2019.01403
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Huang, D. W., Di, R., Wang, J. X., Chu, M. X., He, J. N., Cao, G. L., et al. (2012). Análisis de la secuencia de ADN del gen RFRP de cabra y su posible asociación con la duración media diaria de la insolación. Mol. Biol. Rep. 39, 9167–9177. doi:10.1007/s11033-012-1789-3
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Islam, S., Zeisel, A., Joost, S., La Manno, G., Zajac, P., Kasper, M., et al. (2014). RNA-seq unicelular cuantitativo con identificadores moleculares únicos. Métodos nat. 11, 163–166. doi:10.1038/nmeth.2772
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Kfir, S., Basavaraja, R., Wigoda, N., Ben-Dor, S., Orr, I. y Meidan, R. (2018). Perfil genómico de la maduración del cuerpo lúteo bovino. PLoS One 13 (3), e0194456. doi:10.1371/journal.pone.0194456
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Kim, D., Langmead, B. y Salzberg, S. L. (2015). Hist: Un alineador empalmado rápido con bajos requisitos de memoria. Métodos 12 (4), 357–360. doi:10.1038/nmeth.3317
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Kozomara, A., Birgaoanu, M. y Griffiths-Jones, S. (2019). miRBasa: de las secuencias de microARN a la función. Ácidos nucleicos res. 47 (D1), D155–D162. doi:10.1093/nar/gky1141
Resumen de PubMed | Texto completo de CrossRef | Google Académico
La, Y., Tang, J., He, X., Di, R., Wang, X., Liu, Q., et al. (2019). Identificación y caracterización de mRNAs y lncRNAs en el útero de ovejas Han de cola pequeña politómicas y monótonas (Ovis aries). PeerJ 7, e6938. doi:10.7717/peerj.6938
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Lai, F. N., Zhai, H. L., Cheng, M., Ma, J. Y., Cheng, S. F., Ge, W., et al. (2016). Escaneo del genoma completo para el rasgo de tamaño de camada asociado a genes y SNP bajo selección en cabra lechera (Capra hircus). 6, págs. 1–12. doi:10.1038/srep38096
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Li, Q., Pangas, S. A., Jorgez, C. J., Graff, J. M., Weinstein, M. y Matzuk, M. M. (2008). Funciones redundantes de SMAD2 y SMAD3 en células de la granulosa ovárica in vivo. Mol. Cell. Biol. 28, 7001–7011. doi:10.1128/MCB.00732-08
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Li, Y., Zhang, L., Shang, L., Wang, H., Zou, H., Zhang, H., et al. (2010). Los polimorfismos genéticos en tres loci del gen PRLR y FSHR se correlacionan con el tamaño de la camada en la cabra china Haimen. J. Anim. Vet. Adv. 9, 2835–2838. doi:10.3923/javaa.2010.2835.2838
Li, Z., Wang, J., Zhao, Y., Ma, D., Zhao, M., Li, N., et al. (2021a). scRNA-seq de células de la granulosa del folículo ovárico de diferentes cabras de fertilidad revela patrones de expresión distintos. Reprod. Domest. Anim. 56, 801–811. doi:10.1111/rda.13920
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Li, S., Wang, J., Zhang, H., Ma, D., Zhao, M., Li, N., et al. (2021b). El perfil del transcriptoma de la foliculogénesis caprina revela la interacción de las células de ovocito y granulosa en correlación con diferentes poblaciones de fertilidad. 11 (1), 15698. doi:10.1038/s41598-021-95215-z
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Li, Y., Xu, X., Deng, M., Zou, X., Zhao, Z., Huang, S., et al. (2021c). Identificación y análisis comparativo de ARN largos no codificantes en ovarios de cabra de alta y baja fecundidad durante el estro. Frente. Jineta. 12, 648158. doi:10.3389/fgene.2021.648158
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Liao, Y., Smyth, G. K. y Shi, W. (2014). featureCounts: un eficiente programa de propósito general para asignar lecturas de secuencia a características genómicas. Bioinformática 30 (7), 923–930. doi:10.1093/bioinformatics/btt656
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Liu, S., Chen, S. y Zeng, J. (2018). Señalización de TGF-β: Un papel complejo en la tumorigénesis (revisión). 17, 699–704. doi:10.3892/mmr.2017.7970
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Lotia, S., Montojo, J., Dong, Y., Bader, G. D. y Pico, A. R. (2013). Tienda de aplicaciones Cytoscape. Bioinformática 29, 1350-1351. doi:10.1093/bioinformatics/btt138
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Love, M. I., Huber, W. y Anders, S. (2014). Estimación moderada del cambio de pliegue y dispersión para datos de RNA-seq con DESeq2. Genoma Biol. 15 (12), 1–21. doi:10.1186/s13059-014-0550-8
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Matsuda, F., Inoue, N., Manabe, N. y Ohkura, S. (2012). Crecimiento folicular y atresia en ovarios de mamíferos: Regulación por supervivencia y muerte de células de la granulosa. J. Reprod. 58, 44–50. doi:10.1262/jrd.2011-012
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Miao, X., Luo, Q. y Qin, X. (2016a). El análisis del transcriptoma de todo el genoma en los ovarios de dos cabras identifica genes expresados diferencialmente relacionados con la fecundidad. Gen 582 (1), 69–76. doi:10.1016/j.gene.2016.01.047
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Miao, X., Luo, Q., Zhao, H. y Qin, X. (2016b). Análisis de todo el genoma de miRNAs en los ovarios de cabras Jining Grey y Laiwu Black para explorar la regulación de la fecundidad. 6 (1), 37983. doi:10.1038/srep37983
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Mohammadabadi, M. (2020). Expresión del gen ESR1 en cabra Raini Cashmere mediante PCR en tiempo real. Agric. Biotechnol. J. 12, 177–192. doi:10.22103/JAB.2020.15791.1229
Monniaux, D., Baril, G., Laine, A. L., Jarrier, P., Poulin, N., Cognié, J., et al. (2011). Hormona antimülleriana como marcador endocrino predictivo para la producción embrionaria en la cabra. Reproducción 142, 845–854. doi:10.1530/REP-11-0211
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Mori, T., Amano, T. y Shimizu, H. (2000). Roles de la comunicación de unión gap de células cúmulos en la maduración citoplasmática de ovocitos porcinos cultivados in vitro. Biol. Reprod. 62, 913–919. doi:10.1095/biolreprod62.4.913
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Mossahebi-Mohammadi, M., Quan, M., Zhang, J. S. y Li, X. (2020). Vía de señalización FGF: Un regulador clave de la pluripotencia de las células madre. Frente. Celda. Dev. Biol. 8, 79. doi:10.3389/fcell.2020.00079
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Naserkheil, M., Ghafouri, F., Zakizadeh, S., Pirany, N., Manzari, Z., Ghorbani, S., et al. (2022). La integración multiómica y el análisis de redes revelan posibles genes centrales y mecanismos genéticos que regulan la mastitis bovina. Curr. Temas Mol. Biol. 44 (1), 309–328. doi:10.3390/cimb44010023
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Ozmen, O., Seker, I., Ertugrul, O., Ozkan, E. y Tekin, N. (2011). Receptor de prolactina (<i> PRLR</i>) polimorfismo genético en las razas de ovejas Chios, White Karaman y Awassi. Arch. Anim. Raza. 54, 381–390. doi:10.5194/aab-54-381-2011
Pagel, P., Kovac, S., Oesterheld, M., Brauner, B., Dunger-Kaltenbach, I., Frishman, G., et al. (2005). La base de datos MIPS de interacción proteína-proteína de mamíferos. Bioinformática 21 (6), 832–834. doi:10.1093/bioinformática/bti115
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Pramod, R., Sharma, S., Singhi, A., Pan, S. y Mitra, A. (2013). La morfometría ovárica diferencial y la expresión folicular de BMP 15, GDF 9 y BMPR 1B influyen en la prolificidad en cabra. Reprod. Domest. Anim. 48, 803–809. doi:10.1111/rda.12165
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Qiu, M., Quan, F., Han, C., Wu, B., Liu, J., Yang, Z., et al. (2013). Efectos de las células de la granulosa sobre la esteroidogénesis, proliferación y apoptosis de células estromales y células teca derivadas del ovario caprino. J. Steroid Biochem. Mol. Biol. 138, 325–333. doi:10.1016/j.jsbmb.2013.06.005
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Quan, Q., Zheng, Q., Ling, Y., Fang, F., Chu, M., Zhang, X., et al. (2019). Análisis comparativo de genes expresados diferencialmente entre los ovarios de cabras gestantes y no preñadas utilizando RNA-Seq. J. Biol. Res. Thessal. 26, 3–12. doi:10.1186/s40709-019-0095-9
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Reimand, J., Arak, T., Adler, P., Kolberg, L., Reisberg, S., Peterson, H., et al. (2016). g: Profiler: un servidor web para la interpretación funcional de listas de genes (actualización de 2016). Ácidos nucleicos res. 44, 83–89. doi:10.1093/nar/gkw199
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Reyhan, V. D., Sadeghi, M., Miraei-Ashtiani, S. R., Ghafouri, F., Kastelic, J. P. y Barkema, H. W. (2022). El transcriptoma integrado y los análisis de redes reguladoras identifican genes candidatos y vías que modulan la fertilidad de la oveja. Gene Rep. 28, 101659. doi:10.1016/j.genrep.2022.101659
Sadeghi, M., Bahrami, A., Hasankhani, A., Kioumarsi, H., Nouralizadeh, R., Abdulkareem, S. A., et al. (2022). lncRNA-miRNA-mRNA ceRNA network involved in sheep prolificacy: An integrated approach. Genes. 13 (8), 1295. doi:10.3390/genes13081295
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Salmena, L., Poliseno, L., Tay, Y., Kats, L. y Pandolfi, P. P. (2011). Una hipótesis de ceRNA: ¿La piedra rosetta de un lenguaje de ARN oculto? Celda. 146 (3), 353–358. doi:10.1016/j.cell.2011.07.014
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Saraiva, M., Celestino, J., Araújo, V., Chaves, R., Almeida, A., Lima-Verde, I., et al. (2011). Expresión del receptor de la hormona foliculoestimulante (FSHR) en folículos ováricos de cabra y el impacto del medio de cultivo secuencial en el desarrollo in vitro de folículos preantrales caprinos. Cigoto 19, 205–214. doi:10.1017/S0967199410000511
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Saucedo, L. J., y Edgar, B. A. (2007). Llenando la vía del hipopótamo. Nat. Rev. Mol. Cell. Biol. 8, 613–621. doi:10.1038/nrm2221
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: Un entorno de software para modelos integrados de redes de interacción biomolecular. Genoma Res. 13 (11), 2498–2504. doi:10.1101/gr.1239303
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Sharma, R., Ahlawat, S., Maitra, A., Roy, M., Mandakmale, S. y Tantia, M. (2013). Polimorfismo del gen BMP4 en razas caprinas indias que difieren en prolificidad. Gen 532, 140–145. doi:10.1016/j.gene.2013.08.086
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Sherman, B. T., y Lempicki, R. A. (2009). Análisis sistemático e integrador de grandes listas de genes utilizando recursos bioinformáticos DAVID. Nat. Protoc. 4, 44–57. doi:10.1038/nprot.2008.211
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: Redes de asociación proteína-proteína con mayor cobertura, que apoyan el descubrimiento funcional en conjuntos de datos experimentales de todo el genoma. Ácidos nucleicos res. 47, 607–613. doi:10.1093/nar/gky1131
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Volders, P. J., Anckaert, J., Verheggen, K., Nuytens, J., Martens, L., Mestdagh, P., et al. (2019). LNCipedia 5: Hacia un conjunto de referencia de ARN humanos largos no codificantes. Ácidos nucleicos res. 47 (D1), D135–D139. doi:10.1093/nar/gky1031
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Wang, W., Zhang, E. y Lin, C. (2015). MicroRNAs en la angiogénesis tumoral. Life Sci. 136, 28–35. doi:10.1016/j.lfs.2015.06.025
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Wang, X., Yang, Q., Zhang, S., Zhang, X., Pan, C., Chen, H., et al. (2019). Efectos genéticos de los polimorfismos de un solo nucleótido en el gen GDF9 de la cabra sobre la prolificidad: ¿verdadero o falso positivo? Animales 9, 886. doi:10.3390/ani9110886
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Wei, S., Du, M., Jiang, Z., Hausman, G. J., Zhang, L. y Dodson, M. V. (2016). ARN largos no codificantes en la regulación de la adipogénesis: nuevos ARN arrojan luz sobre la obesidad. Cell. Mol. Life Sci. 73, 2079–2087. doi:10.1007/s00018-016-2169-2
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Xu, Y., Chen, J., Yang, Z. y Xu, L. (2019). Identificación de perfiles de expresión de ARN en cáncer de tiroides para construir una red de ARN endógeno (ceRNA) competidora de ARNm, ARN largos no codificantes (lncRNAs) y microRNAs (miRNAs). Med. Sci. Monit. 25, 1140–1154. doi:10.12659/MSM.912450
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Yang, J., Li, X., Cao, Y. H., Pokharel, K., Hu, X. J., Chen, Z. H., et al. (2019). La expresión comparativa de ARNm y miARN en muflón europeo (Ovis musimon) y ovejas (Ovis aries) proporciona nuevos conocimientos sobre los mecanismos genéticos para el éxito reproductivo femenino. Herencia 122 (2), 172–186. doi:10.1038/s41437-018-0090-1
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Yao, X., Li, F., Wei, Z., Ei-Samahy, M. A., Feng, X., Yang, F., et al. (2022). Análisis integrativo de metilol y transcriptoma de ADN de todo el genoma de ovarios de ovejas hu con alta y baja prolífica. Frente. Celda. Dev. Biol. 10, 820558. doi:10.3389/fcell.2022.820558
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zhang, C., Liu, Y., Huang, K., Zeng, W., Xu, D., Wen, Q., et al. (2011). La asociación de dos polimorfismos de nucleótido único (SNP) en el gen de la hormona del crecimiento (GH) con el tamaño de la camada y la respuesta de superovulación en razas caprinas. Genet. Mol. Biol. 34, 49–55. doi:10.1590/S1415-47572010005000110
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zhang, S., Xu, H., Liu, X., Yang, Q., Pan, C., Lei, C., et al. (2017). El paisaje del transcriptoma del desarrollo muscular de la cabra ovariectomizada. R. Soc. Open Sci. 4, 171415. doi:10.1098/rsos.171415
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zhang, X., Yan, H., Wang, K., Zhou, T., Chen, M., Zhu, H., et al. (2018a). Cabra CTNNB1: perfil de expresión de ARNm de empalme alternativo en testículo y análisis de asociación con el tamaño de la camada. Gen 679, 297–304. doi:10.1016/j.gene.2018.08.061
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zhang, R. Q., Lai, F. N., Wang, J. J., Zhai, H. L., Zhao, Y., Sun, Y. J., et al. (2018b). Análisis de los loci de SNP alrededor de los sitios de inicio de transcripción relacionados con el rasgo de fecundidad de cabra basado en la resecuenciación del genoma completo. Gen 643, 1–6. doi:10.1016/j.gene.2017.12.002
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zhao, Y., Zhang, P., Ge, W., Feng, Y., Li, L., Sun, Z., et al. (2020). Los oligosacáridos de alginato mejoran el desarrollo de las células germinales y el microambiente testicular para rescatar la espermatogénesis interrumpida por busulfano. Teranósticos 10 (7), 3308–3324. doi:10.7150/thno.43189
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zhou, W. Y., Cai, Z. R., Liu, J., Wang, D. S., Ju, H. Q. y Xu, R. H. (2020). ARN circular: Metabolismo, funciones e interacciones con proteínas. Mol. Cáncer 19, 1–19. doi:10.1186/s12943-020-01286-3
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zi, X. D., Hu, L., Lu, J. Y., Liu, S. y Zheng, Y. C. (2020). Comparación de las secuencias y niveles de expresión de genes relacionados con el desarrollo folicular y la atresia entre razas caprinas prolíficas y no prolíficas. Vet. Med. Sci. 6, 187–195. doi:10.1002/vms3.225
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zonaed Siddiki, A., Miah, G., Islam, M., Kumkum, M., Rumi, M. H., Baten, A., et al. (2020). Recursos Genómicos de Cabra: La búsqueda de genes asociados con sus rasgos económicos. Int. J. Genómica. 2020, 5940205–5940213. doi:10.1155/2020/5940205
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Zwick, E., Hackel, P. O., Prenzel, N. y Ullrich, A. (1999). El receptor EGF como transductor central de sistemas de señalización heterólogos. Tendencias Pharmacol. 20, 408–412. doi:10.1016/S0165-6147(99)01373-5
Resumen de PubMed | Texto completo de CrossRef | Google Académico
Cita: Ghafouri F, Sadeghi M, Bahrami A, Naserkheil M, Dehghanian Reyhan V, Javanmard A, Miraei-Ashtiani SR, Ghahremani S, Barkema HW, Abdollahi-Arpanahi R y Kastelic JP (2023) Construcción de una red reguladora de circRNA-lincRNA-lncRNA-miRNA-mRNA identifica genes y vías relacionadas con la fertilidad de las cabras. Frente. Jineta. 14:1195480. doi: 10.3389/fgene.2023.1195480
Recibido: 28 de marzo de 2023; Aprobado: 03 Julio 2023;
Publicado: 21 julio 2023.
Editado por:
Hamayun Khan, Universidad de Agricultura, Pakistán
Revisado por:
Xiukai Caao, Universidad de Yangzhou, China
Ijaz Ahmad, Universidad de Agricultura, Pakistán
Derechos de autor © 2023 Ghafouri, Sadeghi, Bahrami, Naserkheil, Dehghanian Reyhan, Javanmard, Miraei-Ashtiani, Ghahremani, Barkema, Abdollahi-Arpanahi y Kastelic. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia de Atribución Creative Commons (CC BY).
*Correspondencia: Abolfazl Bahrami, a.bahrami@ut.ac.ir; Mostafa Sadeghi, sadeghimos@ut.ac.ir
†Estos autores han contribuido igualmente a este trabajo
Renuncia: Todas las afirmaciones expresadas en este artículo son únicamente las de los autores y no representan necesariamente las de sus organizaciones afiliadas, o las del editor, los editores y los revisores. Cualquier producto que pueda ser evaluado en este artículo o reclamo que pueda ser hecho por su fabricante no está garantizado ni respaldado por el editor.
Date de alta y recibe nuestro 👉🏼 Diario Digital AXÓN INFORMAVET ONE HEALTH
Date de alta y recibe nuestro 👉🏼 Boletín Digital de Foro Agro Ganadero
Noticias animales de compañía
Noticias animales de producción
Trabajos técnicos animales de producción
Trabajos técnicos animales de compañía